|
Erkrankungen von Leber, Gallenwegen,
Pankreas |
|
| Prof. Dr. Hans Huchzermeyer |
| 1.
Erkrankungen der Leber Tab. 1.1 Lebererkrankungen während der Schwangerschaft
Die Lebermitbeteiligung im Rahmen einer schweren Hyperemesis gravidarum stellt keine Lebererkrankung im engeren Sinne dar. Diese Leberfunktionsstörungen als Folge von Mangelernährung, Störungen des Wasser- und Elektrolythaushalts und von metabolischer Ketoazidose bessern sich nach Sistieren des Erbrechens spontan innerhalb weniger Tage (s. Kap. 1.2.1.1. und 4.2.3.). Auf die früher gebräuchliche Klassifizierung hepatobiliärer Erkrankungen in der Schwangerschaft in einen Ikterus in graviditate und einen Ikterus e graviditate sollte heute verzichtet werden. Nicht das fakultative Symptom des Ikterus, sondern die Lebererkrankung selbst sollte in den Vordergrund gerückt werden. Verlässliche Aussagen über Prävalenz und Inzidenz von Lebererkrankungen in der Schwangerschaft liegen nicht vor. Mit einem Fall pro 500 bis 5000 Schwangerschaften scheint ein derartiges Zusammentreffen relativ selten zu sein (69, 70, 74, 75, 77, 152). 1.1. Physiologische Veränderungen Die erhöhten Anforderungen, die eine ungestörte Schwangerschaft an die Leber als zentrales Stoffwechselorgan stellt, werden von einer gesunden Leber ohne Einschränkung bewältigt und auch bei hepatobiliären Erkrankungen ist die Leistungsreserve des Organs in den meisten Fällen noch groß genug, um der funktionellen Mehrbelastung zu genügen. Allerdings zeigen Struktur und Funktion der Leber in der normalen Schwangerschaft bevorzugt zum Schwangerschaftsende einige physiologische Veränderungen, deren Kenntnis Voraussetzung ist für die Erhebung krankhafter Zustände. Leber- und Milzgröße bleiben während der Gravidität unverändert. Da die Leber gegen Schwangerschaftsende durch den Uterus nach oben, hinten und rechts verdrängt wird, ist sie zu diesem Zeitpunkt ebenso wie die Milz schwierig zu palpieren. Ab der 10. Schwangerschaftswoche nehmen zirkulierendes Blutvolumen (30 – 50 %), kardiales Schlagvolumen (30 – 35 %) und zentralvenöser Druck zu, dagegen sinkt der periphere Gefäßwiderstand. Es vergrößert sich vor allem das Plasma-, weniger das Erythrozytenvolumen. Diese Hypervolämie ist nicht nur bei Herz-, sondern auch bei Lebererkrankungen von Bedeutung. Sie führt zu Veränderungen von Blutwerten (Erythrozyten, Hämoglobin, Protein etc.), vor allem trägt sie aber im Zusammenhang mit der interstitiellen Flüssigkeitszunahme bei Leberzirrhose zur Aszitesbildung sowie bei portaler Hypertension zur Auslösung von Ösophagusvarizenblutungen bei. Im Rahmen dieses hyperdynamen Syndroms bilden sich als Folge der vermehrten peripheren Zirkulation, weniger als Folge einer endokrin bedingten Vasodilatation ab dem 2. Schwangerschaftsmonat bei über 60 % der Frauen Leberhautzeichen (Teleangiektasien, Palmarerythem etc.) aus. Sie nehmen im Laufe der Schwangerschaft an Stärke zu und bilden sich nach der Entbindung zurück (69, 70, 74). Trotz der zusätzlichen Belastung des Leberstoffwechsels durch die Schwangerschaft und trotz der Zunahme des Blutvolumens und des Herzzeitvolumens bleibt die Leberdurchblutung im Wesentlichen innerhalb der normalen Schwankungsbreite. Die Berechnung des Leberstromvolumens mittels der Bromsulphthalein (BSP)-Clearance bzw. der Indocyaningrün (ICG)-Clearance zeigt keine Veränderungen der Durchblutung. Die Durchblutung beträgt im Mittel 1554 ml/min/1,73 m² im Vergleich zu Nichtschwangeren mit 1548 ml/min/1,73 m². Auch zwischen den einzelnen Trimestern lassen sich, bei einer allerdings geringen Zahl von Messungen, keine signifikanten Durchblutungsänderungen feststellen. In der Spätschwangerschaft erhält die Leber durchschnittlich 28 % des Blut- und Schlagvolumens, wogegen bei nichtschwangeren Frauen 35 % die Leber erreichen (Tab. 1.2) (61, 69, 70, 74, 126). Die (Farb-) Duplexsonographie der Lebergefäße ist gerade auch in der Schwangerschaft wechselnden Limitationen unterworfen, so dass vergleichbare Normwerte bisher nicht vorliegen. In einer vergleichenden Studie an 67 gesunden Schwangeren zwischen der 10. und 40. Schwangerschaftswoche und 22 gleichaltrigen nichtschwangeren Frauen lag bei den Nichtschwangeren das arterielle bei 0,57 + 0,31 l/min, das portalvenöse bei 1,25 + 0,46 l/min und das Gesamt-Blutflussvolumen der Leber bei 1,82 + 0,63 l/min. Ab der 28. Woche trat eine signifikante Zunahme des portalvenösen und des Gesamt-Blutflussvolumens ein, wohingegen das arterielle Flussvolumen unverändert blieb. (128). Tab. 1.2 Leberdurchblutung in der normalen Schwangerschaft (BSP-Clearance) (126)
Mit zunehmender Schwangerschaftsdauer kann eine Reduktion des Lebervenenflusses beobachtet werden mit allmählicher Normalisierung postpartal. In einer Studie an 75 gesunden Schwangeren änderte sich in der mittleren Lebervene bei 64 % ab der 20. Woche das normale triphasische zu einem monophasischen Flussprofil. Ab der 30. Woche war nur noch bei 8 % der Schwangeren das Flussmuster normal triphasisch (157). In einer weiteren Studie konnte gezeigt werden, dass sich diese Änderung des Lebervenenflusses im Wochenbett zunehmend normalisierte. 6 bis 8 Wochen post partum wiesen 60 % von 30 Untersuchten wieder ein normales Flussprofil in der mittleren Lebervene auf (139). Histologisch bietet die Schwangerschaftsleber ein normales Bild. Elektronenoptisch finden sich jedoch bei ca. drei Vierteln der Schwangeren im letzten Trimenon mitochondriale Veränderungen in Form von Vergrößerungen, Verformungen und kristallinen Einschlüssen. Es dürfte sich um adaptive Veränderungen handeln, wahrscheinlich bedingt durch die erhöhten Steroidhormonspiegel, denen kein Krankheitswert zukommt und die sich nach der Geburt vollständig zurückbilden (Abb. 1.1) (69, 70, 74). Eine besondere Ausprägung erfahren diese strukturellen Alterationen der Mitochondrien im Rahmen der intrahepatischen Schwangerschaftscholestase (s. Kap. 1.3.1.), möglicherweise sind sie auch pathogenetisch bedeutsam bei der akuten Schwangerschaftsfettleber (s. Kap. 1.3.3).
Abb. 1.1 Ausschnitt einer Leberparenchymzelle bei ungestörter Schwangerschaft 8. Monat. Neben wenigen, normal strukturierten Mitochondrien finden sich zahlreiche vergrößerte Mitochondrien mit kristallinen Innenstrukturen und dichtem Cristae-Besatz. Bis auf eine geringe Vakuolisierung des endoplasmatischen Retikulums sind die übrigen Zellorganellen normal. Vergr. 13800fach.
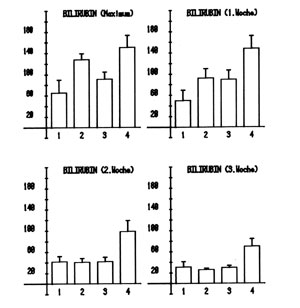
Tab. 1.3 fasst die typischen laborchemischen Veränderungen während der Schwangerschaft zusammen. Die Bestimmung von Enzymaktivitäten im Serum ist auch in der Schwangerschaft für die Diagnose und Differenzierung von hepatobiliären Erkrankungen von entscheidender Bedeutung. Während die Aktivitäten von GOT(AST), GPT (ALT), GLDH und LDH im Serum während der gesamten Schwangerschaft im Normbereich verbleiben, nimmt die Aktivität der CHE geringfügig ab und kann in der zweiten Schwangerschaftshälfte leicht erniedrigte Aktivitäten erreichen. Die Aktivität der γ-GT, die bereits bei gesunden jungen, nichtschwangeren Frauen um etwa 20 – 30 % niedriger ist als bei Männern, sinkt ab dem 2. Trimenon ab und ist in der Spätschwangerschaft signifikant niedriger als bei gleichaltrigen nichtschwangeren Frauen. Selbst im Rahmen der akuten Virushepatitis findet sich dieser hormonale Effekt auf die γ-GT bei jüngeren Frauen im Gegensatz zu Frauen in der Postmenopause oder zu Männern. Nicht nur zu Beginn, sondern auch im Longitudinalverlauf sind die Werte der Kollektive 1 bis 3 signifikant niedriger als die der Kollektive 4 bis 6 (Abb. 1.2).
Abb. 1.2 Aktivität der γ-GT im Serum (Normbereich < 18 U/l) bei akuter Virushepatitis zum Zeitpunkt des GPT-Maximums sowie in der 1., 2. und 3. Krankheitswoche. (1) Schwangere mit fortgeschrittener Schwangerschaft. (2) Frauen mit Einnahme von Ovulationshemmern in höherer Dosierung bis zum Erkrankungsbeginn. (3) Frauen bis 40 Jahre (ohne 1 und 2). (4) Frauen über 45 Jahre. (5) Männer bis 40 Jahre. (6) Männer über 40 Jahre. Damit verhält sich die γ-GT grundsätzlich anders als die übrigen Cholestase anzeigenden Enzyme 5’-Nucleotidase, AP und LAP, die während der Schwangerschaft ansteigen, und ist damit ungleich besser zur Diagnostik hepatobiliärer Erkrankungen als diese Enzyme geeignet. Der kontinuierliche Anstieg der AP auf das Zwei- bis Dreifache der Norm ab dem dritten Monat ist durch das plazentare Isoenzym der AP bedingt, der ebenfalls ab dem dritten Monat nachzuweisende LAP-Anstieg auf das Drei- bis Achtfache ist auf die in der Plazenta gebildete Oxytocinase zurückzuführen. Die Aktivität der 5’-Nucleotidase steigt im 2. und 3. Trimenon nur gering an (7, 69, 70, 74). Der Bilirubin-Spiegel bleibt im Normbereich, obwohl die mütterliche Leber ab dem vierten Monat das fetale Bilirubin zusätzlich zu eliminieren hat. Dies ist vor allem auf die Hämodilution zurückzuführen. Auch die Gesamt-Gallensäuren sind normal (< 10 µmol/l). Alpha-Fetoprotein (AFP) wird in der fetalen Leber synthetisiert und sezerniert. AFP ist zum Ende des 2. Monats im mütterlichen Serum nachzuweisen, steigt bis zur 31. SSW im Mittel auf Werte über 200 ng/ml an, um dann bis zur Geburt leicht abzusinken. Höhere Werte werden bei fetalen Mißbildungen (z. B. Anenzephalie, Spina bifida) oder beim hepatozellulären Karzinom der Mutter gefunden (s. Kap. 1.2.11). Die Zunahme der Triglyzerid- und Cholesterinkonzentrationen sowie der drei Haupt-Lipoproteinfraktionen im Serum ist physiologisch und dient der Versorgung des Feten wie der plazentaren Hormonsynthese. Von klinischer Relevanz ist der Anstieg der Konzentration der Faktoren VII und X um 80 %, des Faktors VIII um 100 % und des Faktors I (Fibrinogen) um 70 %, gleichzeitig ist die Fibrinolyse herabgesetzt. Ansteigende Werte von Thrombin-Antithrombinkomplex (TAT), Prothrombinfragment 1 und 2 sowie D-Dimer weisen auf die Aktivierung der Gerinnung hin. Die resultierende Hyperkoagulabilität wird allerdings in der normalen Schwangerschaft durch die Expansion des Plasmavolumens kompensiert. Bei elektrophoretischer Trennung der Plasmaproteine findet sich eine Abnahme der Albumine (bis 50 rel. %) und eine Zunahme der Alpha- und besonders der Betaglobuline, während die Gammaglobuline unverändert bleiben oder gering abnehmen. Das Gesamteiweiß nimmt bis zum Ende der Schwangerschaft durchschnittlich um 0,5 g/100 ml ab, nur in Einzelfällen werden hypoproteinämische Werte erreicht (7, 69, 70, 74). Mit Hilfe von Anamnese, klinischen, klinisch-chemischen, sonographischen und Dopplersonographischen Untersuchungen, nur in ausgewählten Fällen ergänzt durch die Leberbiopsie, sollte es heute fast immer gelingen, die Ätiologie einer hepatobiliären Erkrankung während einer Schwangerschaft zu klären (Tab. 1.4). Tab. 1.4 Ursachen einer Leberfunktionsstörung während der verschiedenen Schwangerschaftsstadien
1.2 Schwangerschaftsunabhängige Lebererkrankungen 1.2.1. Akute und chronische Virushepatitiden 1.2.1.1. Akute Virushepatitis Die akute Neuinfektion mit den Hepatitis-Viren A, B, C, D und E definiert die akute Virushepatitis. Der Infektion mit dem GB-Virus C (Hepatitis G-Virus) kommt wahrscheinlich nur eine geringe klinische Bedeutung zu. Die Häufigkeit der akuten Hepatitisinfektionen in Deutschland liegt bei etwa 35 pro 100.000 Einwohner und Jahr. In der Schwangerschaft ist die akute Virushepatitis mit etwa 40 % die häufigste Ursache einer Lebererkrankung. Die Erkrankungszahlen sind mit der Häufigkeit der Hepatitis in der Allgemeinbevölkerung vergleichbar. Die Schwangerschaft birgt kein erhöhtes Risiko, an einer Virushepatitis zu erkranken, und auch die Suszeptibilität gegenüber Hepatitisviren ist in den verschiedenen Phasen der Schwangerschaft nicht unterschiedlich. Die Diagnose einer akuten Virushepatitis bietet auch in der Schwangerschaft keine spezifischen Probleme. Die typischen klinisch-chemischen Befunde, insbesondere die Konstellation des Enzymmusters und die virologischen Parameter, erlauben sicher die Diagnose und Differenzierung z.B. von anderen Ikterusformen in der Schwangerschaft. Allerdings ist die Bestimmung der Aktivitäten von AP, 5’-Nucleotidase und LAP, die physiologisch im Verlauf der Schwangerschaft allmählich ansteigen, zur Erkennung einer cholestatischen Verlaufsform weniger geeignet. Stets sollte daher die Aktivität der γ-GT bestimmt werden, die nicht nur die Cholestase anzeigt, sondern auch ein Hinweis für die klinische Ausheilung der Hepatitis sein kann. Offenbar unter dem Einfluss der Östrogene und/oder des Progesterons sind die Werte der γ-GT im Mittel niedriger als bei vergleichbaren Kontrollen, besonders in der 2. Hälfte der Schwangerschaft. Dieses besondere Verhalten der Aktivität der γ-GT im Serum sei an 4 verschiedenen Kollektiven mit akuter Virushepatitis A oder B, und zwar zum Zeitpunkt des GPT-Maximums sowie in der 1., 2. und 3. Krankheitswoche im Vergleich zu den Aktivitäten von AP und LAP verdeutlicht (Abb. 1.3). Das erste Kollektiv umfasst Hepatitiserkrankungen bei Schwangerschaften im 2. und 3. Trimenon, das zweite Frauen mit Einnahme oraler Kontrazeptiva bis zum Beginn der akuten Hepatitis, das dritte bis zur Hepatitiserkrankung gesunde Frauen bis 40 Jahre und das 4. Kollektiv ebenfalls bis zur Hepatitiserkrankung gesunde Frauen über 45 Jahre. Der hormonale Einfluss auf die γ-GT ist in den drei ersten Kollektiven stets erkennbar, die Werte sind signifikant niedriger als beim 4. Kollektiv, den Frauen nach der Geschlechtsreife. Der durch die Schwangerschaft bedingte Aktivitätsanstieg lässt sich besonders am Verhalten der LAP zeigen, wohingegen deutlich erhöhte Werte der AP durch den Beitrag der Plazenta-AP erst im 3. Trimester zu finden sind. Eine Verminderung der γ-GT-Aktivität bzw. normale Aktivitäten können auch während Schwangerschaften bei der benignen rekurrierenden intrahepatischen Cholestase (s. Kap. 1.2.4.), bei der autoimmunen Hepatitis und anderen Autoimmunerkrankungen (s. Kap. 1.2.6.) bei der intrahepatischen Schwangerschaftscholestase (s. Kap. 1.3.1.) sowie beim HELLP-Syndrom (s. Kap. 1.3.2.) beobachtet werden (69, 70, 74). Da die Anfangsphase einer akuten Hepatitis mit gastrointestinalen Symptomen wie Inappetenz, Übelkeit und Erbrechen einhergehen kann, muss differentialdiagnostisch das Vorliegen von Emesis bzw. Hyperemesis gravidarum bedacht werden (s. Kap. 4.2.3.) (73). Das Enzymmuster (Referenzbereich in Klammern)
einer 25-jährigen Erstgravida in der 12. Woche mit seit 14
Tagen bestehender Hyperemesis zeigt als Folge der toxischen Leberzellschädigung
eine über das vierfache des oberen Referenzwertes erhöhte
Aktivität der
Der unverhältnismäßig hohe Anstieg der mitochondrialen vorzugsweise perivenös lokalisierten GLDH im Vergleich zur zytosolischen GPT und bilokulären GOT weist darüber hinaus darauf hin, dass die toxische Läsion überwiegend den zentrolobulären Bereich betrifft. Weiterhin ist bei der Wertung des Enzymmusters bei einer akuten
Hepatitis während der Schwangerschaft auf die physiologische
Depression der CHE-Aktivität (ab 2. Trimenon) hinzuweisen,
die allein nicht als Hinweis auf einen besonders schweren oder protrahierten
Verlauf der Hepatitis angesehen werden darf.
Unabhängig von der Art des Erregers werden Verlauf, Krankheitsdauer und Ausheilung der Erkrankung – zumindest in Westeuropa – durch die Schwangerschaft nicht negativ beeinflußt. Es muß jedoch mit Rückwirkungen der Infektion auf die Frucht gerechnet werden (69, 70, 74, 75, 91, 119, 179). Die auf den gesamten Zeitraum der Schwangerschaft berechnete Frühgeburtenrate beträgt etwa 16 %, beim Auftreten der Virushepatitis in der Spätschwangerschaft erhöht sich diese auf 29 % und gleichzeitig findet sich eine 10 %ige Totgeburtenrate. Bisher ist keines der Hepatitisviren in einen ursächlichen Zusammenhang mit dem Auftreten von Missbildungen zu bringen (Tab. 1.5) (69, 70, 74, 75). Tab. 1.5 Kindliche und mütterliche Prognose bei Lebererkrankungen während der Schwangerschaft
Die Infektion des Kindes ist somit als das wesentlichste Risiko einer Virushepatitis während der Schwangerschaft anzusehen. Eine verlässliche Aussage zur Übertragung der verschiedenen Erreger von der Mutter auf das Kind ist derzeit für das Hepatitis A-, B- und C-Virus möglich (Tab. 1.6). Tab. 1.6 Hepatitisviren und Schwangerschaft
Bei einer akuten Hepatitis A während der Schwangerschaft ist eine Übertragung des Virus auf das Neugeborene bei intakter Plazenta kaum zu erwarten. Vielmehr werden die mütterlichen HAV-Antikörper auf das Kind übertragen, die ihm für die ersten 10 – 12 Lebensmonate Immunität gegen Hepatitis A verleihen. Eine HAV-Exposition bewirkt dann eine aktive Immunisierung der Säuglinge und zwar ohne die Symptome einer klinischen Infektion. Insgesamt tritt eine akute Hepatitis A beim Neugeborenen direkt nach der Geburt selten auf und zwar nur dann, wenn die Mutter gleichzeitig akut perinatal oder während der Stillperiode erkrankt. Durch passive Immunprophylaxe kann das Neugeborene geschützt werden. Für die aktive Immunisierung des Neugeborenen mit einer inaktivierten Virusvakzine bzw. für die aktiv-passive Immunisierung liegen ausreichende Erfahrungen bisher nicht vor. Gleiches gilt für die Wirkung der aktiven Immunisierung auf schwangere oder stillende Frauen. Es sollte nur bei eindeutiger Indikation aktiv geimpft werden (Tab. 1.6) (37, 69, 70, 74, 77). Im Gegensatz zur Hepatitis A kommt der Infektion der Neugeborenen HBV-positiver Mütter die größte Bedeutung zu, da die Kinder häufig zu chronischen Virusträgern werden. Eine Infektionsgefährdung des Kindes ist in unterschiedlichem Ausmaß bei akuter Hepatitis B und beim chronischen HBV-Trägertum der Mutter gegeben. Bei einer akuten Hepatitis B kurz vor oder nach der Geburt werden über 90 % der Kinder infiziert, beim Auftreten im ersten und zweiten Trimenon weniger als 10 %. Offensichtlich verhindert eine intakte Plazenta die Übertragung. Sowohl bei einer Hepatitis B in der Spätschwangerschaft als auch bei chronischen HBs-Ag-Trägerinnen erfolgt die Infektion überwiegend perinatal, wobei die höchste Inzidenz dieser vertikalen Übertragung im wesentlichen von der Menge des zirkulierenden HBV bestimmt wird. Entsprechend signalisiert der Nachweis von HBe-Antigen und von viraler DNS ein hohes Infektionsrisiko von etwa 90 % und zwar unabhängig von den verschiedenen geographischen Regionen. In der deutschen Bevölkerung beträgt die Häufigkeit von HBV-Dauerträgern zwischen 0,3 bis 0,8 %, die Inzidenz bei den bei uns lebenden Ausländern ist mit 8 % deutlich höher. Entsprechend wird in Deutschland bei etwa 800.000 Geburten pro Jahr die Zahl der Neugeborenen, die jährlich durch das HBV gefährdet werden, auf mindestens 2.500 bis 6.500 geschätzt. Abhängig vom Ausmaß der Virämie bei der Mutter schwankt das Infektionsrisiko dieser Kinder zwischen 10 und über 90 %. Von den HBV-infizierten Kindern entwickeln nur wenige innerhalb von 6 Monaten post partum das Vollbild einer akuten Hepatitis. 80 – 90 % der Kinder werden dagegen zu chronischen Virusträgern. Die Infektion verläuft asymptomatisch und anikterisch, nur die HBs-Ag-Persistenz und meist leicht erhöhte Aktivitäten der Transaminasen im Serum weisen auf die stattgehabte HBV-Infektion hin. Offensichtlich durch die unterschiedliche Immunreaktion des Wirtsorganismus bestimmt, entwickelt sich dagegen bei Kindern im Schulalter in 25 – 50 % und bei Erwachsenen in etwa 10 % eine chronische Infektion. Die jährlich mindestens 750 – 1.500 chronischen HBV-Infektionen bei Neugeborenen in Deutschland stellen nicht nur ein konstantes Virusreservoir dar, 20 – 30 % von ihnen sterben im Jugend- oder Erwachsenenalter an Zirrhosen oder hepatozellulären Karzinomen. Als Folge der perinatalen Infektion sind demnach pro Jahr 200 – 400 Todesfälle zu erwarten. Um die vertikale Transmission des HBV zu unterbinden, ist ein generelles Schwangerenscreening auf HBs-Ag nach der 32. Schwangerschaftswoche und die passiv-aktive Impfung aller Kinder HBs-Ag-positiver Mütter gleich nach der Geburt erforderlich. Diese Maßnahmen dürften nicht nur zu einer effektiven Unterbrechung der Infektionskette führen, sondern erscheinen auch nach den Erfahrungen aus anderen Ländern volkswirtschaftlich als sehr sinnvoll. Da die Infektion in der Regel unter der Geburt stattfindet, ist die passiv-aktive Impfung aller Kinder HBs-Ag-positiver Mütter unmittelbar post partum mit Auffrischimpfungen nach 4 Wochen und 6 Monaten erforderlich. Die Immunisierung verhindert in bis zu 95 % der Fälle eine Infektion, allerdings kann es bei sehr hohen HBV-DNA-Konzentrationen trotz Impfung zur vertikalen Transmission kommen (69, 70, 74, 146, 152, 177). Möglicherweise ist es günstig, bei HBs-Ag-positiven Müttern mit hoher Virämie diese durch eine Therapie mit Lamivudin im letzten Schwangerschaftsmonat zu reduzieren, um das Risiko eines Impfmisserfolgs beim Kind zu verringern (203). Zur weiteren Evaluierung dieses Therapieansatzes sind größere kontrollierte Studien notwendig. Bei der hohen Effektivität auch einer ausschließlich aktiven Schutzimpfung erscheint es sinnvoll, zukünftig die allgemeine Impfung von Neugeborenen und Kleinkindern zu empfehlen. Die Schwangere, die einem hohen Risiko einer HBV-Infektion ausgesetzt ist, kann nach den bisherigen Erfahrungen gefahrlos aktiv immunisiert werden, da der zur Verfügung stehende inaktivierte Hepatitis-B-Impfstoff keine Virämie erzeugt. Ist ein sofortiger Impfschutz erforderlich, empfiehlt sich die kombinierte aktiv-passive Immunisierung. Gleichzeitig schützt eine solche HBV-Prophylaxe gegen Hepatitis D (Tab. 1.6). Auch das Hepatitis C-Virus wird wie das HBV überwiegend durch Blut, Blutprodukte und Sekrete übertragen. Die innerfamiliäre oder sexuelle Übertragung scheint relativ selten zu sein, besonders im Vergleich zur HBV-Infektion. Das Infektionsrisiko der Neugeborenen liegt bei 4 – 6 %, wobei dieses Risiko mit dem Titer der HCV-RNA bei der Mutter korreliert. Eine hohe Transmission des HCV findet sich bei Kindern von HIV-Infizierten oder anderen Risikogruppen. Wie bei der Hepatitis B entwickeln die infizierten Kinder in über 80 % eine chronische Infektion, selten heilt die Hepatitis C spontan aus. Entsprechend dürfte auch hier nach 20 bis 30 Jahren das Risiko der Entstehung eines hepatozellulären Karzinoms und einer Leberzirrhose erhöht sein. Zur Prophylaxe der Hepatitis C existieren derzeit nur die Möglichkeiten der Expositionsprophylaxe (69, 70, 74, 154, 204). Berichte über eine vertikale Transmission des Hepatitis D-Virus liegen nur vereinzelt vor. Voraussetzung ist das gleichzeitige Vorliegen einer HBV-Infektion. Anti-HDV-Antikörper werden passiv dem Feten übertragen und verschwinden innerhalb von drei Monaten. Eine Immunprophylaxe gegen HBV verhindert auch eine perinatale Infektion mit HDV. Sichere Aussagen zur Infektionskette des GB-Virus C (HGV/GBV-C), das ebenfalls wie das HCV zur Familie der Flaviviridae gehört, lassen sich augenblicklich noch nicht machen. Möglicherweise ist das Infektionsrisiko der Neugeborenen höher als bei einer HCV-Infektion, besonders begünstigt wird die Transmission offensichtlich auch hier bei HIV-Infizierten und anderen Risikogruppen. Wahrscheinlich ist das Risiko einer persistierenden Infektion bei HGV-RNA positiven Kindern erhöht, Zeichen einer Lebererkrankung finden sich jedoch nicht (134). Insgesamt dürften Infektionen mit HDV und GBV-C, aber auch mit HEV in Westeuropa keine größere Rolle für die perinatale Hepatitis spielen. Die akute Hepatitis E, zumeist endemisch und epidemisch in den Staaten der 3. Welt auftretend, wird analog zum HAV vor allem fäko-oral übertragen. Während die Hepatitis A vor allem Kinder und Jugendliche befällt, findet sich eine Häufung der Hepatitis E bei Erwachsenen im 2. bis 4. Lebensjahrzehnt. In Endemiegebieten werden gerade bei schwangeren Frauen im letzten Trimenon fulminante Verläufe einer Hepatitis E beobachtet mit einer Letalität bis zu 20 %. Im Vergleich zu den anderen Virushepatitiden ist die Abortrate deutlich erhöht und möglicherweise besteht auch eine erhöhte vertikale Transmission mit signifikant erhöhter perinataler Morbidität und Mortalität. Die Ursache für die hohe Komplikationsrate bei Schwangeren ist unklar. Zu diskutieren sind Eigenarten des Virus selbst oder die häufig vorliegenden mangelhaften Ernährungs- und Hygieneverhältnisse. In Europa wird die akute Hepatitis E selten und zumeist nach Auslandsaufenthalten beobachtet (9, 14, 74, 95). Neben den klassischen Hepatitisviren A bis E kommen noch weitere, primär nicht hepatotrope Viren wie Herpes simplex-Viren (HSV), Zytomegalie-Virus (CMV) oder Epstein-Barr-Virus (EBV) für die Ausbildung einer Hepatitis in der Schwangerschaft in Frage. Von diesen Erregern der Herpesgruppe weisen HSV und CMV mit fortschreitender Schwangerschaft eine zunehmende Reaktivierungsrate auf, und sie können wahrscheinlich begünstigt durch eine Verminderung der zellulären Immunität besonders im 3. Trimenon eine fulminante Hepatitis mit hoher mütterlicher und fetaler Letalität (40 – 50 % der Fälle) auslösen (3, 48). Ebenso können bakterielle und parasitäre Infektionen (Leptospiren, Mykobakterien, Treponemen, Amöben, Echinokokken, Plasmodien etc.) zur seltenen Ursache einer Leberschädigung und gelegentlich auch eines Ikterus während der Schwangerschaft werden. Hier ist auch der Ikterus bei schweren bakteriellen Allgemeininfektionen und Sepsis (z. B. septischer Abort, Puerperalsepsis, Urosepsis) zu erwähnen. Ursache des Ikterus ist eine toxische Leberzellschädigung, eine Hämolyse kann hinzutreten. 1.2.1.2. Chronische Virushepatitis Persistiert die Virusinfektion über 6 Monate und weist die Leber des Patienten histologisch ein unterschiedlich ausgeprägtes entzündliches Infiltrat auf, liegt eine chronische Hepatitis vor. Die Infektion mit dem Hepatitis B-Virus verläuft in 5 – 10 %, die Hepatitis C-Infektion in bis zu 80 % der Fälle chronisch. Als inkomplettes, defektes Virus tritt das Hepatitis D-Virus nur gemeinsam mit dem Hepatitis B-Virus auf. Während die Koinfektion in bis zu 90 % ausheilt, verläuft die Superinfektion in der Regel chronisch. Hepatitis A und E führen nicht zu einer chronischen Verlaufsform. Die früher geltende histologische Klassifikation in eine chronisch-persistierende, chronisch-aktive und chronisch-lobuläre Hepatitis ist heute abgelöst worden durch das Konzept des Grading and Staging. Durch Angabe der Grundkrankheit, der Stärke der entzündlichen Aktivität und des Ausmaßes des fibrotischen Umbaus ist es jetzt möglich, die Krankheitsprogression wie auch mögliche Therapieeffekte besser abzuschätzen. Im Vergleich zur akuten Virushepatitis sind Berichte über Schwangerschaften bei chronischen Virushepatitiden seltener. Schwangerschaften bei chronischer Hepatitis mit minimaler, milder oder mäßiggradiger Entzündungsaktivität bzw. Fibrose beeinflussen weder die Lebererkrankung noch sind Komplikationen für die Schwangerschaft zu erwarten, und selbst bei schweren chronischen Hepatitiden mit hoher Entzündungsaktivität und schwerer Fibrose wird in den meisten Fällen die Leberfunktion nicht negativ beeinflußt. Die kindliche Prognose ist dagegen bei einer chronischen Hepatitis schwerer Aktivität durch eine erhöhte Inzidenz von Frühgeburtlichkeit, Mangelentwicklung und perinataler Mortalität eingeschränkt (Tab. 1.5) (66, 69, 70, 74). Bei den virusinduzierten chronischen Hepatitiden haben sich Interferone als therapeutisch wirksam erwiesen. Zwar konnten in Einzelfällen unter der Behandlung mit alpha-Interferon Schwangerschaften normal ausgetragen werden, doch reichen diese geringen Erfahrungen nicht aus, die Monotherapie mit alpha-Interferon zur Behandlung der chronischen viralen Hepatitiden während der Schwangerschaft zu empfehlen. Auch von einer Gabe von Interferon-alpha bei der akuten Hepatitis C, wie es außerhalb der Schwangerschaft wegen der hohen Neigung zur Chronizität diskutiert wird, ist während der Schwangerschaft abzuraten. Kontraindiziert ist die Interferon-Ribavirin-Kombinationstherapie bei chronischer Hepatitis C, da beim Nukleosid-Analogon Ribavirin offensichtlich ein signifikantes teratogenes Risiko für den Feten besteht. Ebenso kann Lamivudin außerhalb von Studien als Option zur Therapie der chronischen Hepatitis B nicht empfohlen werden, da auch bei diesem Nukleosid-Analogon Studien über die Unbedenklichkeit während der Schwangerschaft nicht vorliegen. Wie bei der akuten Virushepatitis B sollten auch die Neugeborenen von Müttern mit einer chronischen Hepatitis B sofort nach der Geburt kombiniert passiv-aktiv immunisiert werden (70, 78). Beim Vorliegen chronischer Virushepatitiden ist nicht nur das materno-fetale Übertragungsrisiko, sondern auch die Möglichkeit einer paterno-fetalen Virustransmission zu berücksichtigen. Bei chronischer HBV-Infektion des Ehemannes ist die Möglichkeit einer Virustransmission durch infizierte Spermien wahrscheinlich gegeben. Bei Vorliegen einer chronischen Hepatitis C des Ehemannes wird zur Risikoreduktion das Verfahren der assistierten Reproduktion mit aufbereiteten Spermien diskutiert (194). 1.2.2. Medikamentöse und toxische Leberschäden Während der Schwangerschaft nehmen zwischen 30 und 80 % aller schwangerer Frauen die verschiedensten Arten und Kombinationen von Pharmaka ein. Dies erfolgt aufgrund einer behandlungsbedürftigen Erkrankung, nicht selten werden aber auch Medikamente in Unkenntnis einer bereits bestehenden Schwangerschaft eingenommen. Zusätzliche Einflüsse können von chemischen Substanzen in der Ernährung, im Haushalt oder in der Industrie ausgehen. Bei derartigen Expositionen wird in erster Linie an Fehlbildungen und bleibende Schäden des Feten gedacht. Die zentrale Rolle der Leber in der Biotransformation von pharmakologischen oder chemischen Substanzen macht aber auch verständlich, daß die verschiedensten Substanzgruppen leberschädigende Wirkungen entfalten können. Man nimmt an, dass in Deutschland etwa 500 – 1000 Pharmaka potentiell hepatotoxisch sein können. Man unterscheidet im Wesentlichen zwei Formen der chemisch-toxischen Leberzellschädigung, die direkt toxische und die idiosynkratische Form. Das Bild einer durch Medikamente und exogene Toxine hervorgerufenen Leberschädigung ist vielgestaltig, jede andere Lebererkrankung kann imitiert werden. Am häufigsten findet sich jedoch klinisch, klinisch-chemisch und histologisch das Bild einer intrahepatischen Cholestase, einer toxischen Hepatitis oder einer Mischform zwischen diesen beiden Reaktionsformen. Daher sollte bei vorliegender oder vermuteter Schwangerschaft bei der Anwendung von Arzneimitteln der zu erwartende Nutzen nicht nur gegen teratogene oder embryotoxische Schäden für die Frucht, sondern auch gegen mögliche Gefahren für die Mutter – d. h. hier besonders im Hinblick auf toxische Leberschäden – abgewogen werden. Das Auftreten einer Leberschädigung ist dabei jedoch nicht an die Schwangerschaft gebunden, und auch die Schwere des Schadens und der klinische Verlauf lassen während der Schwangerschaft keine Besonderheiten erkennen. Überwiegend werden Arzneimittel-induzierte Cholestasen beobachtet, deren Schweregrad und Dauer erheblich variieren können. Hauptsächlich wurde dieses Bild bei Patientinnen mit Hyperemesis gravidarum beobachtet, die mit Chlorpromazinderivaten behandelt wurden. Sehr viel seltener als eine medikamentenbedingte Cholestase ist das Krankheitsbild einer schweren Hepatitis (69, 70, 78). Das Spektrum alkoholischer Lebererkrankungen umfasst die asymptomatische bis symptomarme alkoholische Fettleber, das variantenreiche Krankheitsbild der Alkoholhepatitis und schließlich die alkoholtoxische Leberzirrhose. Eine Gefährdung von Mutter und Kind besteht beim Auftreten einer akuten fulminant verlaufenden Alkoholhepatitis und bei fortgeschrittenen Zirrhosen mit rascher Progredienz und/oder der Verstärkung eines Pfortaderhochdrucks. Das Hauptproblem eines übermäßigen mütterlichen Alkoholkonsums in der Schwangerschaft besteht im Auftreten von Schäden bei Embryo und Feten in Form der fetalen Alkoholeffekte und des selteneren fetalen Alkoholsyndroms mit den typischen klinischen Veränderungen. 1.2.3. Metabolische Lebererkrankungen 1.2.3.1. Morbus Wilson Der Morbus Wilson ist eine seltene autosomal-rezessiv vererbte Kupferspeicherkrankheit mit einer Erkrankung pro 30.000 Lebendgeburten. Das veränderte Wilson-Gen (ATP 7B), das auf Chromosom 13 liegt und von dem bisher über 200 verschiedene Mutationen beschrieben wurden, führt zu einer progredienten Kupferablagerung vor allem in Leber und Gehirn (hepatolentikuläre Degeneration). In der Regel werden die Patienten klinisch symptomatisch im Jugend- oder jungem Erwachsenenalter. Entsprechend der vorherrschenden Symptomatik kann der Morbus Wilson in eine hepatische, eine neurologisch-psychiatrische oder eine gemischte Form klassifiziert werden. Das Bild der hepatischen Manifestation ist vielgestaltig. Es reicht von der inaktiven Hepatitis über die chronisch-aktive Hepatitis bis hin zur Leberzirrhose mit ihren typischen Komplikationen. Als akuter Morbus Wilson wird eine fulminant verlaufende akute Hepatitis verstanden, die in etwa 5 % der Fälle auftritt, häufig mit einer Coombs-negativen akuten Hämolyse assoziiert ist und bei der in der Regel die hochdringliche Lebertransplantation diskutiert werden muß. Betroffen sind meist junge Menschen im 2. oder 3. Lebensjahrzehnt mit einer Bevorzugung des weiblichen Geschlechts gegenüber dem männlichen im Verhältnis 3:1. Nur die frühe Diagnose des Morbus Wilson (Nachweis von erniedrigtem Coeruloplasmin und hoher Kupferausscheidung im Urin, eines Kayser-Fleischer-Kornealrings sowie von extrapyramidal-motorischen Symptomen) und eine konsequente lebenslange Therapie verhindern ein Fortschreiten der Erkrankung. Die Standardtherapie besteht aus der Gabe von Chelatbildnern: D-Penicillamin (900 - 2400 mg/Tag) oder von Trientine (1200 - 2700 mg/Tag) in Kombination mit Zinksalzen. Ausgeprägte sekundäre Organschäden führten früher beim unbehandelten Morbus Wilson zur Einschränkung der Fertilität, so dass Schwangerschaften seltene Ausnahmen darstellten. Eine primäre oder sekundäre Amenorrhö oder wiederholte Spontanaborte, die möglicherweise durch erhöhte Kupferkonzentrationen in intrauterinen Sekreten induziert werden, müssen den Verdacht auf einen Morbus Wilson lenken. Seit Einführung der spezifischen Therapie nimmt die Zahl der Beobachtungen von erfolgreichen und komplikationslosen Schwangerschaften ständig zu: In über 150 Schwangerschaften wurde D-Penicillamin, in über 20 Trientine eingesetzt. Eine Teratogenität der Chelatbildner oder von Zink konnte bisher nicht gesehen werden. Durch den vermehrten Kupferbedarf des Feten und aufgrund des schwangerschaftsspezifischen Anstiegs des Coeruloplasmins kann es zu einer kurzzeitigen Besserung des Krankheitsverlaufs kommen, so dass für die letzten 6 Wochen eine Dosisreduzierung des Penicillamins auf 25 – 50 % der Ausgangsdosis empfohlen wird. Eine adjuvante Gabe von Vitamin B6 (20 – 40 mg/Tag) sollte fortgeführt werden. Die kupferentspeichernde Therapie sollte bei schwangeren Patientinnen jedoch unter keinen Umständen unterbrochen werden, um keine akuten Wilson-Krisen zu riskieren. Die Differentialdiagnose von HELLP-Syndrom und akutem M. Wilson kann im Einzelfall schwierig sein (Tab. 1.5) (70, 74, 78, 118, 131, 176, 181, 192). 1.2.3.2. Hepatische Porphyrien Die hepatischen Porphyrien sind eine heterogene Gruppe von Stoffwechseldefekten, die auf verschiedenen Gendefekten der Enzyme entlang der Hämbiosynthese beruhen. Zu unterscheiden sind akute und chronische hepatische Porphyrien. Die akuten Formen, die überwiegend Frauen betreffen, weisen eine komplexe klinische Symptomatik auf, die insbesondere von intermittierenden Anfällen starker abdomineller Schmerzen und von kardiovaskulären und neuropsychiatrischen Symptomen geprägt ist. Da die akute intermittierende Porphyrie bevorzugt bei Frauen in der zweiten bis vierten Lebensdekade auftritt, werden Schwangerschaften vorwiegend bei dieser autosomal-dominant vererbten Porphyrieform gesehen, während Schwangerschaften bei den anderen hepatischen Porphyrien (89, 96) zu den Seltenheiten gehören. Heute geht man davon aus, dass Schwangerschaften kein erhöhtes Risiko für Mutter und Kind darstellen. In früheren Jahren war man von einer schubprovozierenden und die Erkrankung aggravierenden Wirkung der Schwangerschaft ausgegangen. Dies läßt sich einerseits darauf zurückführen, daß ein nicht unbeträchtlicher Teil dieser Graviden gleichzeitig porphyrinogene Pharmaka, vor allem Barbitursäure-Präparate, eingenommen hatte, andererseits vorzugsweise solche Fälle publiziert wurden, in denen es während der Schwangerschaft zu einer Manifestation oder Exazerbation der akuten Porphyrie gekommen war. Die Wahrscheinlichkeit der Vererbung des Defekts für jedes Kind beträgt 50 % (1, 15, 70, 74, 96, 166). Das Risiko einer Kontrazeption mittels Ovulationshemmern wird unterschiedlich beurteilt. Teils schreibt man ihnen eine krisenauslösende Wirkung, teils eine günstige Beeinflussung der Erkrankung zu. In den seltenen Fällen, wo ein Zusammenhang zwischen Krise und prämenstrueller Phase besteht (ovulozyklische Formen) wird ein Therapieversuch mit oralen Kontrazeptiva empfohlen (70, 74, 149). 1.2.3.3. Hyperlipoproteinämien Die Leber nimmt im Fettstoffwechsel eine zentrale Rolle ein, regelt sie u.a. im Wesentlichen die Produktion und die Elimination der verschiedenen Lipoproteine. Dies macht verständlich, dass Störungen der Leberfunktion auch den Lipidmetabolismus beeinträchtigen und zu komplex veränderten Lipoproteinmustern führen können. Nach ihrer Zusammensetzung werden bei den Lipoproteinen 5 Hauptklassen unterschieden: triglyzeridreiche Chylomikronen und very-low-density-Lipoproteine (VLDL), intermediate-density-Lipoproteine (IDL), cholesterinreiche low-density-Lipoproteine (LDL) sowie für den reversen Cholesterintransport verantwortliche high-density-Lipoproteine (HDL). Die Apolipoproteine (z.B. Apo B-100 assoziiert mit VLDL, IDL und LDL sowie Apo A-1 mit HDL und Chylomikronen) regeln den Transport und den Metabolismus dieser Lipoproteine. Die Hyperlipoproteinämien lassen sich in isolierte Hypercholesterinämien, isolierte Hypertriglyzeridämien und in kombinierte Hyperlipidämien einteilen. Abhängig von der Pathogenese werden primäre von sekundären Hyperlipidämien unterschieden. Die primären Dyslipoproteinämien sind genetisch definiert und haben monogene oder polygene Störungen zur Ursache. Häufiger sind sekundäre Formen als Folge von Erkrankungen (Adipositas, Diabetes mellitus, Hypothyreose, M. Cushing, Lebererkrankungen, nephrotisches Syndrom, Alkoholabusus u.a.) oder einer Pharmakotherapie (Östrogene, anabole Steroide, Glukokortikoide, Thiazide, einige Betablocker u.a.). Allerdings sind die Grenzen zwischen diesen beiden Formen fließend. Eine primär genetische Störung kann durch Ernährungseinflüsse, Medikamente und Erkrankungen manifest oder aggraviert werden. In der normalen Schwangerschaft verändern sich die Lipid- und Apolipoproteinspiegel und steigen signifikant an. Diese Veränderungen beginnen im ersten Trimenon, erreichen ihr Maximum im dritten und normalisieren sich in den ersten 6 Wochen post partum, überwiegend (wie die Triglyzeride) bereits in der ersten Woche. Der Anstieg betrifft vor allem die Triglyzeride im Mittel um das 2- bis 3-fache, das Gesamtcholesterin um das 1,5- bis 2-fache und die Phospholipide um das 1,5-fache, aber auch geringer die freien Fettsäuren und Lipoprotein (a). Der HDL-Cholesterinspiegel steigt zwar bis zur 24. Schwangerschaftswoche an, fällt dann jedoch ab, um bei der Entbindung nur noch 15 % über den Ausgangswerten zu liegen (92, 99, 123). Diese Veränderungen des Fettstoffwechsels lassen sich zum Teil mit der hormonellen Umstellung in der Schwangerschaft erklären. Diskutiert werden Effekte von Östrogenen, Progesteron, Kortisol, Choriongonadotropin, humanem plazentaren Laktogen, Insulin und Glukagon. Die erhöhten Triglyzeridspiegel, bedingt durch vermehrte Produktion und verminderten Katabolismus von VLDL, resultieren im Wesentlichen aus dem Anstieg der Östrogene. In früheren Studien sah man den gleichen Effekt unter oralen Kontrazeptiva. Dosisabhängig erhöhten Östrogene die Triglyzeride in allen Lipoproteinfraktionen, während Gestagene diesem Anstieg entgegenwirkten (49, 99, 200). Bei der Schwangerschaftshyperlipoproteinämie handelt es sich somit um eine sekundäre gemischte Hyperlipidämie. Sie ist als physiologischer Regulationsvorgang sowohl für die adäquate Versorgung des Feten mit einem materno-fetalen Transfer von VLDL und IDL als auch für die Bereitstellung von Bausteinen für die plazentare Hormonsynthese von Bedeutung und damit nicht therapiebedürftig. Eingehende lipidanalytische Untersuchungen sind
in der Schwangerschaft angezeigt bei: Erhöhte Cholesterinspiegel und verminderte Konzentrationen der HDL im Plasma gelten als bedeutsame Risikofaktoren der Arteriosklerose. Für die Annahme, dass mehrfache Schwangerschaften aufgrund der physiologischen kombinierten Hyperlipidämie ein atherogenes Risiko in sich bergen, existieren keinerlei Hinweise. Schwangerschaften bei familiärer Hypercholesterinämie mit Erhöhung von LDL (Typ IIa nach Frederickson) bzw. bei kombinierter Hyperlipidämie mit Erhöhung von LDL und VLDL (Typ IIb) sind in Einzelfällen beschrieben. Angina pectoris und Myokardinfarkte können sich auch in der Schwangerschaft manifestieren. Ausgeprägte Hypertriglyzeridämien (über 1000 mg/dl) aufgrund erhöhter VLDL oder Chylomikronen sind als Ursache von akuten Oberbauchschmerzen oder akuter Pankreatitis von klinischer Relevanz. Pathophysiologisch wird hier eine Verminderung der rheologischen Eigenschaften des Blutes, besonders durch die Chylomikronen, diskutiert. Für unser Thema von größerer Bedeutung sind daher die primären Hypertriglyzeridämien mit Erhöhung von VLDL, Chylomikronen oder Chylomikronen plus VLDL (entsprechend Typ IV, I, V) wie auch die sekundären Formen, die bei hohen Werten mit Hepatosplenomegalie, Bauchschmerzen und akuter Pankreatitis einhergehen können (31, 33, 122). Da Lipidsenker in der Schwangerschaft sehr selten indiziert sind, fehlen Erfahrungen und sie gelten hier wie in der Stillzeit als kontraindiziert. Dennoch rechtfertigt eine erfolgte Medikation nicht einen Schwangerschaftsabbruch. Die bisher vorliegenden Daten seien im Folgenden kurz zusammengefasst. Die Cholesterinsynthese-Enzymhemmer (CSE-Hemmer) zeigen im Tierversuch nur in hoher Dosierung embryotoxische und teratogene Wirkungen. Beim menschlichen Fetus wäre eine Schädigung durch die Synthesehemmung von Cholesterin und anderen Zwischenprodukten der Cholesterinbiosynthese möglich. In retrospektiven Einzelfallberichten und prospektiven Fallsammlungen wurden verschiedene Fehlbildungen beobachtet, jedoch wurden auch zahlreiche Verläufe mit normalem Schwangerschaftsausgang nach Therapie im ersten Trimenon mitgeteilt. Nach gegenwärtigem Kenntnisstand ist derzeit weder für die Statine noch für die anderen Präparate eine statistische Risikoberechnung möglich. Probucol gilt wegen seines nicht völlig geklärten Wirkungsmechanismus und seiner langen Halbwertzeit von bis zu 30 Tagen zu Recht als absolut kontraindiziert. Es muss mindestens ein halbes Jahr vor einer geplanten Schwangerschaft abgesetzt werden. Die wasserunlöslichen Anionenaustauscherharze Colestyramin und Colestipol werden enteral nicht resorbiert, sondern vollständig mit dem Stuhl ausgeschieden. Nur bei sehr hoher Dosierung können eine Fettmalabsorption und ein Mangel an fettlöslichen Vitaminen resultieren. Da bei niedriger Dosis (bis 9 g pro Tag) über mehrere Monate ohne die genannten Nebenwirkungen therapiert werden und eine eventuell notwendige Kompensation durch mittelkettige Triglyzeride und Vitaminsubstitution erfolgen kann, sind aus unserer Sicht Anionenaustauscherharze in der Schwangerschaft als relativ sicher anzusehen. Bei allen Fibraten und deren Analoga ist wegen verminderter fetaler Glucoronidkonjugation besonders im dritten Trimenon mit einer Kumulation zu rechnen. Fehlbildungen sind allerdings weder beim Tier noch beim Menschen bislang berichtet worden. Bei der Nikotinsäure handelt es sich um eine physiologische Substanz mit kurzer Halbwertzeit von 30 Minuten. Bei einer Gabe von 2 g pro Tag ergaben sich bisher keine Anhaltspunkte für eine Toxizität für den Fetus. Trotzdem sollte man in der Schwangerschaft aus Sicherheitsgründen erst bei Triglyzeridwerten über 1000 mg/dl, wenn die Mutter akut gefährdet ist, Nikotinsäure einsetzen. Eine Kombination mit Austauscherharzen dürfte in der Schwangerschaft ebenfalls möglich sein. Eine derartige medikamentöse Therapie ist jedoch erst dann gerechtfertigt, wenn es mit diätetischen Maßnahmen (Fettreduktion auf 15 % der Energiezufuhr, Anbieten von mittelkettigen Triglyzeriden, Reduktion von Zucker, Verbot von Alkohol) nicht gelingt, die Triglyzeridwerte deutlich unter 1000 mg/dl zu senken (77, 110). 1.2.3.4. Hepatische Glykogenosen Die Glykogenspeicherkrankheiten sind genetisch determiniert und werden meistens autosomal-rezessiv vererbt. Defekte können nahezu jedes der im Glykogenmetabolismus involvierten Enzyme betreffen. Die Inzidenz aller Glykogenosen beträgt eine Erkrankung auf 20.000 Lebendgeburten. Durch bessere diagnostische und therapeutische Möglichkeiten erreichen heute viele Patienten das Erwachsenenalter. Auf die folgenden Formen der hepatischen Glykogenosen, die im Wesentlichen durch Hepatomegalie und Hypoglykämien gekennzeichnet sind und die wahrscheinlich keine schwere Einschränkung der Fertilität aufweisen, sei hier hingewiesen (21):
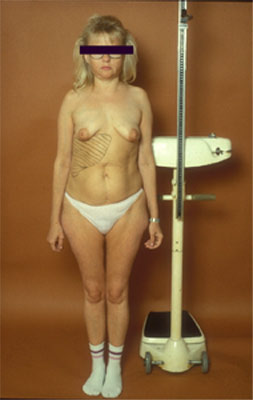
Kinder mit Glykogenosen vom Typ I weisen die folgenden klinischen Merkmale auf: Neigung zu Hypoglykämien, erhöhte Konzentrationen von Laktat, Harnsäure, Cholesterin und Triglyzeriden sowie eine Wachstumsverzögerung. Der seltenere Subtyp Ib (etwa 10 % der Fälle) kann zusätzlich rezidivierende bakterielle Infektionen infolge Neutropenie und Neutrophilendysfunktion ebenso wie chronisch entzündliche Darmerkrankungen entwickeln. Die Bildung von hepatischen Adenomen in der zweiten und dritten Lebensdekade sowie Nierenerkrankungen (Proteinurie, Steinbildung, Hypertonie) gehören zu den Spätkomplikationen. Kinder mit Typ III-Glykogenosen weisen klinisch ein ähnliches Bild wie Typ I-Glykogenosen auf (Hepatomegalie, Minderwuchs, Hypoglykämie, Hyperlipidämie, Wachstumsretardierung). Beim Subtyp IIIa (85 % der Patienten) finden sich zusätzlich zur Leberbeteiligung Myopathien von Skelett- und Herzmuskulatur. Beim Subtyp IIIb ist ausschließlich die Leber beeinträchtigt. Gleichzeitige Nierenerkrankungen weisen Typ III-Patienten nicht auf. Durch eine effektive Therapie lässt sich die Prognose der betroffenen Kinder verbessern (häufige kohlenhydratreiche Mahlzeiten aus Maisstärke, bei Typ I Restriktion der Fruktose und Galaktoseaufnahme sowie Gicht-Intervallbehandlung etc.), so dass heute die überwiegende Zahl das Erwachsenenalter erreichen. Bei Typ III-Glykogenosen verschwinden die hepatischen Symptome postpubertär, die Muskelschwäche ist dagegen bei Typ IIIa progredient. Die Pubertät tritt meistens verspätet ein. Bei der hormonalen Kontrazeption ist auf Ethinylestradiol zu verzichten aufgrund der Bedeutung in der Pathogenese von Hyperlipidämie und Leberadenomen. Obwohl die Fertilität anscheinend nicht wesentlich beeinträchtigt ist (Frauen mit Typ III-Glykogenose weisen relativ häufig eine polyzystische Degeneration der Ovarien auf), ist das Zusammentreffen von hepatischen Glykogenosen und Schwangerschaft ausgesprochen selten. Die Hauptgefährdung für Mutter und Kind gehen von Hypoglykämien, die strenger diätetischer Maßnahmen bedürfen, aus, aber auch von der Entwicklung einer Niereninsuffizienz (Typ I) oder einer Kardiomyopathie (Typ III) bei der Mutter. Anstelle des Urikostatikums Allopurinol zur Harnsäureelimination sollte dem in der Schwangerschaft sichereren Probenecid der Vorzug gegeben werden. Fallberichte von erfolgreichen Schwangerschaften sowohl bei Typ Ia wie bei Typ IIIa wurden publiziert. Sogar nach kombinierter Nieren- und Lebertransplantation bei Glykogenose Typ Ia konnte eine Schwangerschaft erfolgreich ausgetragen werden (21, 27, 39, 77, 86, 101, 105, 115, 164). Eine eigene Beobachtung einer 1957 geborenen Patientin mit Glykogenose Typ Ia weist in typischer Weise einen proportionierten Minderwuchs (150 cm, 43 kg), ein Puppengesicht und eine Hepatomegalie auf (Abb. 1.4). Als Kind fiel die Patientin bereits durch Hepatomegalie, Minderwuchs, Fettpolster im Bereich der Wangen, niedrige Blutzuckerspiegel ohne klinische Abb. 1.4 Glykogenose Typ Ia. 30-jährige Patientin mit proportioniertem Kleinwuchs und Puppengesicht. Hepatomegalie ohne Splenomegalie. Symptomatik, Hypertriglyzeridämie (bis 1314 mg/dl), mäßiggradige Hypercholesterinämie (bis 260 mg/dl) und Anämie auf. Diätetische Maßnahmen erfolgten nicht. Erst 1970 wurde histologisch-enzymatisch das Vorliegen einer Glykogenose Typ Ia gesichert, seither häufige kohlenhydratreiche Kost. 1978 wurde die rechte Niere bei chronischer Pyelonephritis und bei Vorliegen von Harnsäuresteinen operativ entfernt. 1979 und 1984 mussten perianale Fisteln operativ versorgt werden, seit 1980 traten intermittierend Durchfälle auf, ohne dass hier nach Ursachen gefahndet wurde. Die 1. Schwangerschaft im Jahre 1983 wurde bei drohender Plazentainsuffizienz 2 Wochen vor dem Termin in der 39. Schwangerschaftswoche durch Kaiserschnitt beendet. Das 2270 g schwere unreife männliche Neugeborene verstarb 2 Tage nach der Geburt an einem schweren Atemnotsyndrom. Die Obduktion ergab keinen Hinweis auf eine Glykogenose. Die 2. Schwangerschaft ein Jahr später wurde Ende der 40. Schwangerschaftswoche ebenfalls durch Sectio beendet, gleichzeitig erfolgte die Sterilisatio. Das 3030 g schwere weibliche Neugeborene war gesund. Während der 2. Schwangerschaft fanden sich folgende Laborwerte (Normalwerte in Klammern): Hb 103 g/l (120 – 160 g/l) 1986 wurde ein Morbus Crohn mit Befall von terminalem Ileum, Coecum
und Rektum endgültig gesichert. Noch 1986 erfolgte die Ileocoecalresektion
mit Ileoascendostomie bei Konglomerattumor und Abszessbildung. Wiederholte
Operationen von Analfisteln und –abszessen bei anorektalem
Morbus Crohn führten zur Zerstörung des Kontinenzorgans.
Des Weiteren bestehen jetzt Leberrundherde (bisher keine histologische
Sicherung der vermuteten Adenome), eine Cholezystolithiasis, eine
Osteoporose und eine Nephropathie der verbliebenen linken Niere
mit rezidivierenden Infekten bei Harnsäuresteinen. Bei dem 1963 geborenen Bruder der Patientin besteht ebenfalls eine Glykogenose Typ Ia (152 cm, 45 kg), auch hier wurde die Diagnose 1970 gesichert. Eine ausgeprägte Hypoglykämieneigung, das Auftreten einer akuten Pankreatitis bei Triglyzeridwerten zwischen 1380 und 2150 mg/dl und der Nachweis von Leberrundherden im Jahre 1987 kennzeichnen den bisherigen klinischen Verlauf. Anzumerken ist, dass eine weitere 1955 geborene Schwester im 9. Lebensmonat verstarb bei Hepatomegalie und niedrigen Nüchternblutzuckerwerten, das zwei Brüder des Großvaters väterlicherseits und ein Bruder des Großvaters mütterlicherseits als Säuglinge bzw. als Kleinkinder an unbekannter Krankheit verstarben. Typ VI-Glykogenosen sind ausgesprochen selten im Gegensatz zum x-chromosomal vererbten Subtyp der Typ IX-Glykogenosen. Der Verlauf beider Typen ist symptomarm, Hepatomegalie und Wachstumsretardierungen verschwinden postpubertär (21). Da die Erwachsenen praktisch symptomlos sind, ist dies vielleicht eine Erklärung dafür, dass Schwangerschaften bei diesen „benignen“ Glykogenosen m. W. bisher nicht publiziert wurden. Keine Glykogenose im eigentlichen Sinne ist die Typ 0-Glykogenose. Vielmehr führt ein Defekt der Glykogensynthetase zu einer verminderten Glykogenspeicherung. Die bei Kindern auftretenden Hypoglykämien und Hyperketonämien verschwinden im Erwachsenenalter. Lediglich im Rahmen von Schwangerschaften können erneut Hypoglykämien beobachtet werden (21). 1.2.3.5. Hereditäre Hämochromatose Die hereditäre Hämochromatose ist durch eine erhöhte intestinale Eisenresorption mit konsekutiver Eisenablagerung in verschiedenen Geweben, besonders in Leber, Pankreas, Herz und endokrinen Organen, charakterisiert. Es ist die häufigste angeborene Stoffwechselerkrankung mit autosomal-rezessivem Erbgang. Aufgrund der Eisenverluste durch Menstruation und Schwangerschaft manifestiert sich die Erkrankung bei Frauen fünf- bis zehnmal seltener als bei Männern. Auch ein Hypogonadismus – bei Männern meist als Spätsymptom auftretend – findet sich selten bei Frauen, da die Organschädigung durch die Eisenablagerung erst nach der Menopause einsetzt. Lediglich eine ausgeprägte Eisenüberladung kann auch bei jüngeren Frauen zu einer primären oder sekundären Amenorrhoe führen. Während eine Aderlasstherapie (Zielkriterium sind Serum-Ferritinwerte von 50 – 100 ng/ml) den Hypogonadismus bei Männern häufig nicht bessert, stellen Aderlässe bei Frauen mit krankheitsbedingter Amenorrhoe eine wirksame Therapie dar. Somit besitzt diese Erkrankung nur in Ausnahmefällen eine geburtshilfliche Bedeutung (13, 34, 40). 1.2.3.6. Morbus Gaucher Die nicht-neuropathische Form kann sich vom Kindes– bis zum Erwachsenenalter manifestieren mit großen Unterschieden in Schweregrad und Befall der inneren Organe. Bei symptomatischen Patienten finden sich häufig eine Hepatosplenomegalie, eine Infiltration des Knochenmarks mit Anämie und Thrombozytopenie sowie ein Abbau des Knochengerüsts. Oft liegen geringe Leberfunktionsstörungen vor, ein Leberversagen ist jedoch selten. Bis in die sechziger Jahre des vergangenen Jahrhunderts wurde in der Annahme, dass eine Schwangerschaft ein zu hohes Risiko für die Gaucher-Patientin darstelle, von einer Schwangerschaft abgeraten und ein therapeutischer Abort sowie eine Sterilisation empfohlen (69, 186). Diese Auffassung wurde in der Folgezeit mit der Verbesserung der Behandlungsmaßnahmen allmählich revidiert und vielleicht ergeben sich mit den neuen Strategien des Enzymersatzes bzw. der Substrathemmung auch für die Schwangerschaft weitere erfolgreiche Therapieformen. Bei etwa zwei Dritteln der Patientinnen setzt die Pubertät verzögert ein, die Fertilität ist dann jedoch nicht gestört. Die Spontanabortrate ist möglicherweise leicht erhöht (53, 69, 160). Während der Schwangerschaft bringt die Hepatomegalie keine Probleme mit sich, die Leberfunktion ist nur selten leicht gestört. Komplikationen ergeben sich eher aus der Beteiligung anderer Organe. So wurden Schwangerschaften in Kombination mit Osteonekrosen, mit Blutungskomplikationen bei Thrombozytopenie und Anämie, mit Herzinsuffizienz bei Herzmuskelbeteiligung sowie mit Antiphospholipid-Syndrom beschrieben (22, 23, 53, 175, 186). Als das Hauptproblem ist eine Panzytopenie anzusehen, die aus der Milzvergrößerung und der Knochenmarkverdrängung durch die Gaucher-Zellen resultiert. Früher wurde in Einzelfällen eine Splenektomie durchgeführt, ebenso wurden Kortikosteroide erfolgreich eingesetzt (186). Seit über 10 Jahren steht nun eine gut wirksame Enzymersatztherapie mit Imiglucerase und Alglucerase zur Verfügung. Die bisherigen Erfahrungen an wenigen Schwangeren mit unterschiedlichen Symptomen sind vielversprechend, fetotoxische Effekte ergaben sich bisher nicht (38, 41, 175). Das Vorliegen eines Morbus Gaucher oder die Behandlung mittels Enzymsubstitution rechtfertigen u. E. keinen risikobegründeten Abbruch der Schwangerschaft. Für die neue Therapieoption, die Synthese der Glucocerebroside durch Glucosylceramid-Synthase-Hemmstoffe (z.B. Miglustat) zu reduzieren, fehlen bisher jegliche Erfahrungen bei Schwangeren. 1.2.4. Hereditäre Cholestasen Eine heterogene Gruppe von Cholestase-Syndromen stellen die familiären Formen der intrahepatischen Cholestase dar. Sie zeichnen sich durch Defekte im Transport von Gallensäuren und Gallelipiden aus. Da der Beginn der Cholestase im Säuglings- und Kindesalter liegt, mit rascher Ausbildung einer Zirrhose, fasst man diese Lebererkankungen unter dem Begriff der progressiven familiären intrahepatischen Cholestase (PFIC) zusammen. Beim PFIC Typ 1 (Bylers disease bzw. Byler syndrome) ließ sich der Defekt auf dem ATP8B1 (FIC-1)-Gen, beim PFIC Typ 2 (Byler like syndrome) auf dem ABCB11 (BSEP)-Gen und beim PFIC Typ 3 auf dem ABCB4 (MDR-3)-Gen nachweisen. Gallensäurensynthesedefekte können ebenfalls zu einer familiären Form der neonatalen Cholestase führen. Differentialdiagnostisch sind vor allem bei den Typen PFIC-1 und PFIC-2 die häufigeren Cholestase-Formen Alagille-Syndrom (defektes Gen Jagged 1) und extrahepatische Gallengangsatresie zu berücksichtigen. Die Therapie der Kinder mit PFIC Typ 1 und 2 besteht in der partiellen biliären Diversion (Cholezystoenterokutaneostomie), mit Typ 3 in der Gabe von Ursodesoxycholsäure, bei Therapieversagen wird die Lebertransplantation erforderlich. Diese kindlichen Cholestaseformen mit Progression zur Leberzirrhose besitzen für unser Thema praktisch keine Bedeutung im Gegensatz zu zwei milde verlaufenden Varianten, die sich oft erst im Erwachsenenalter manifestieren. Die intrahepatische Schwangerschaftscholestase (ICP) und/oder die Bildung von Cholesteringallensteinen lassen sich der PFIC-3 (diese Erkrankungen werden in Kapitel 1.3.1. und 2. besprochen), die benigne rekurrierende intrahepatische Cholestase (BRIC) der PFIC-1 zuordnen (8, 12, 17, 58, 76, 130, 138, 187). Allerdings prädisponieren ABCB4-Mutationen nur bei bis zu 15 % der Schwangeren zur ICP. Die klinische Heterogenität der ICP macht daher wahrscheinlich, dass auch weitere defekte Gene wie FIC1 und BSEP involviert sind. Ebenso ist eine Beeinflussung von Transportproteinen über Kernrezeptoren durch die erhöhten Konzentrationen von Östrogenen, Progesteron und Kortikosteroiden zu diskutieren (Tab. 1.7) (17, 44, 76, 90, 138, 168, 171, 187). Tab. 1.7 Charakteristika der hereditären Cholestasen
Die BRIC (Summerskill-Walshe-Tygstrup-Syndrom) ist ein rezidivierendes Cholestase-Syndrom, hervorgerufen durch verschiedene Mutationen im ATP8B1 (FIC1)-Gen. Trotz oft extremem Juckreiz bildet sich keine Zirrhose aus und es besteht auch nicht die Gefahr der Entwicklung eines hepatozellulären Karzinoms. Oft treten die Schübe nach Infektionen auf, um dann nach einer Dauer von 3 – 4 Monaten spontan abzuklingen. Klinisch-chemisch finden sich im Schub eine Hyperbilirubinämie, erhöhte Gallensäurenkonzentrationen, normale oder leicht erhöhte Aktivitäten von GOT und GPT und eine mäßiggradige Erhöhung der AP, jedoch bei normaler Aktivität der γ-GT. Histologisch besteht eine blande Cholestase ohne Entzündungszeichen, die ERCP zeigt normale Gallenwege. Bisher wurden erst wenige Schwangerschaften bei diesem Syndrom berichtet, wobei beweisende molekulargenetische Untersuchungen auf entsprechende Mutationen fehlen. Fast immer löste die Schwangerschaft (wie auch orale Antikonzeptiva) frühzeitig einen Schub mit Pruritus (und Ikterus) aus, der nach der Entbindung abklang, jedoch auch über längere Zeit persistieren konnte. Therapeutisch werden sowohl Colestyramin zur Linderung des Pruritus als auch Ursodeoxycholsäure (UDC) zur Minderung des fetalen Risikos jedoch mit unsicherem Erfolg gegeben. Charakteristisch sind – wie erwähnt - normale Aktivitäten der γ-GT im Serum. Ist die γ-GT erhöht, müssen andere Mutationen, z.B. ein ABCB4-(MDR3)-Defekt in Betracht gezogen werden (58, 70, 76, 83, 138). Es ist zu vermuten, daß durch den vermehrten Einsatz molekularer Diagnostik weitere hereditäre Cholestaseformen in der Schwangerschaft charakterisiert werden. So dürfte dem Bericht über eine rekurrierende familiäre intrahepatische Schwangerschaftscholestase, assoziiert mit einer chronischen Lebererkrankung, bei vier Schwestern wahrscheinlich eine eigene neue Krankheitsentität zugrunde liegen (102). 1.2.5. Funktionelle Hyperbilirubinämien Nicht zu den Cholestase-Syndromen zählen die genetischen Hyperbilirubinämien, die autosomal-rezessiv vererbte Defekte des Transports, der Speicherung und des Metabolismus von Bilirubin aufweisen. Sie sind jedoch von differentialdiagnostischer Relevanz. Dem Morbus Gilbert-Meulengracht und dem Crigler-Najjar-Typ-II-Syndrom liegt ein unterschiedlich ausgeprägter Mangel der Bilirubin-UGT-Aktivität zugrunde. Der Morbus Gilbert-Meulengracht wird in den meisten Fällen durch eine Schwangerschaft nicht beeinflusst. Die unkonjugierte Hyperbilirubinämie kann sich aber auch in der Schwangerschaft manifestieren, verstärken oder sogar zurückbilden. Die Schwangerschaft verläuft ungestört. Es besteht keine Assoziation mit dem HELLP-Syndrom (205). Über Schwangerschaften bei dem sehr seltenen Crigler-Najjar-Syndrom Typ II (Arias-Syndrom) wurde bisher in Einzelfällen berichtet. Diese im Vergleich mildere Form einer unkonjugierten Hyperbilirubinämie mit benignem Verlauf spricht auch in der Schwangerschaft gut auf eine Phototherapie und/oder die Verabreichung von Phenobarbital (nicht während der Embryogenese) an. Ein Neugeborenenikterus führt nicht zu einem toxischen Hirnschaden. Dem Dubin-Johnson-Syndrom wie dem Rotor-Syndrom liegen Transportstörungen organischer Anionen an der kanalikulären Hepatozytenmembran zugrunde. Im Falle des Dubin-Johnson-Syndroms findet sich eine Mutation des MRP2-Proteins, beim Rotor-Syndrom besteht neben einer reduzierten biliären Exkretionskapazität zusätzlich eine deutliche Reduktion der hepatozellulären Bilirubinspeicherkapazität (138). Eine Schwangerschaft wie auch die Gabe östrogenhaltiger Antikonzeptiva verstärken beim Dubin-Johnson-Syndrom in den meisten Fällen den vorbestehenden Ikterus oder lassen bis dahin asymptomatische Fälle manifest werden. Der Ikterus kann zu jedem Zeitpunkt der Schwangerschaft akzentuiert werden oder sogar erst nach der Entbindung auftreten. Nach der Entbindung bildet sich der Ikterus allmählich in 1 – 2 Wochen zurück. Ausreichende Erfahrungen über den Verlauf von Schwangerschaften beim Rotor-Syndrom liegen bisher nicht vor (25, 28, 29, 70, 174). Der Gallensäuretransport ist bei diesen genetischen Hyperbilirubinämien nicht gestört, ein Pruritus tritt nicht auf. Eine Therapie ist nicht verfügbar. Da nach bisherigem Kenntnisstand kein erhöhtes Risiko für Mutter oder Kind besteht, ist die Indikation zur Interruptio nicht gegeben. 1.2.6. Autoimmune Hepatitis (AIH) Die autoimmune Hepatitis, die primär biliäre Zirrhose und vermutlich auch die primär sklerosierende Cholangitis werden den autoimmunen Lebererkrankungen zugeordnet. Von einem Überlappungssyndrom spricht man, wenn die AIH zusätzlich die Symptome und Marker einer anderen autoimmunen Lebererkrankung aufweist. Die AIH ist als chronisch progrediente Hepatitis definiert, die in der Regel mit Hypergammaglobulinämie und zirkulierenden Autoantikörpern einhergeht mit hohem Risiko, langfristig in eine Zirrhose überzugehen. Ätiologie und Pathogenese der AIH sind unbekannt. Neben Virusinfektionen als ursächlicher Faktor der Autoimmunität wird eine genetische Prädisposition vermutet. Die Prävalenzraten betragen zwischen 3 und 17 pro 100.000 Einwohner. Die AIH ist Bestandteil des Syndroms der chronischen Hepatitis. Vor einer definitiven Diagnose müssen daher virale, metabolische, hereditäre, medikamentös-toxische und cholestatische Leberkrankheiten differentialdiagnostisch ausgeschlossen werden. In etwa einem Viertel der Fälle imponiert die AIH zu Beginn der Erkrankung als akute Hepatitis, häufiger ist jedoch ein schleichender Beginn. In der Folge führen schubweise Verläufe zu einer wechselnden klinischen Symptomatik entsprechend einer chronischen Hepatitis bzw. einer Zirrhose. Extrahepatische Autoimmunsyndrome wie Autoimmunthyreopathie, Crest-Syndrom, rheumatoide Arthritis, thrombozytopene Purpura, Colitis ulcerosa etc. können assoziiert sein. Aufgrund des Autoantikörperprofils und des klinischen Bildes werden 3 Typen unterschieden. Die AIH-Typ I ist mit 80 % die häufigste Form. 70 % der Erkrankten sind Frauen mit einem Altersmaximum zwischen 16 und 30 Jahren. 30 % weisen assoziierte immunologische Erkrankungen auf. Typisch ist das Auftreten von Antikörpern gegen Kerne (ANA) und/oder gegen glatte Muskulatur (SMA). In den meisten Fällen ist der klinische Verlauf schleichend mit uncharakteristischen Symptomen, 25 % haben aber bei Diagnosestellung bereits eine Zirrhose. Die AIH-Typ II ist seltener als der Typ I (in Europa 20 %). Auch hier ist das weibliche Geschlecht bevorzugt, das Altersmaximum liegt im Kindesalter um das 10. Lebensjahr. Im Vergleich zu Typ I ist der klinische Verlauf häufiger akut, ebenso ist die Zahl extrahepatischer Immunsyndrome größer. Immunserologisch finden sich Antikörper gegen Mikrosomen aus Leber und Nieren (Anti-LKM1). Der seltenste Typ ist die AIH-Typ III. Über 90 % der Erkrankten sind Frauen, das Altersmaximum liegt zwischen dem 20. und 40. Lebensjahr. Dieser Typ wird im Serum durch Antikörper gegen lösliches Antigen aus Leber und Pankreas (Anti-SLA/LP) gekennzeichnet. Häufig finden sich zusätzliche andere Autoantikörper wie SMA und AMA. Die Standardtherapie bei allen Formen der AIH besteht, unabhängig vom klinischen Bild oder vom Autoantikörperprofil, in der Gabe von Kortikosteroiden mit oder ohne Azathioprin. Die Monotherapie mit Kortikosteroiden oder die Kombination aus Steroiden und Azathioprin sind gleich effektiv. Die Dauer der immunsuppressiven Therapie muß individuell beantwortet werden. Bei einem Teil der Patienten können nach 2 bis 3 Jahren zunächst die Steroide, dann Azathioprin ausgeschlichen werden. Unter einer solchen Therapie liegt heute die 10-Jahres-Überlebensrate über 90 %, die 20-Jahres-Überlebensrate über 80 %, ohne Immunsuppression verlief die AIH dagegen früher häufig tödlich. Berichte über Schwangerschaften bei AIH sind bisher relativ selten, zudem stammen verläßlichere Daten erst aus den letzten Jahren. Denn vor etwa einem Jahrzehnt war es erstmals durch das Studium der vielschichtigen Klinik sowie der zellulären und molekularen Immunpathologie gelungen, die AIH als eigenständige Entität zu klassifizieren. In den vorangegangenen Jahrzehnten war die AIH in die ätiologisch heterogene Gruppe der chronischen Hepatitiden bzw. Leberzirrhosen subsumiert worden (182). Da die AIH vorwiegend das weibliche Geschlecht im gebärfähigen Alter betrifft, wird man zukünftig vermehrt mit dem Zusammentreffen dieses Krankheitsbildes mit einer Schwangerschaft rechnen müssen. Dies um so mehr, da unter einer adäquaten immunsuppressiven Therapie bei nur gering entzündlicher Aktivität wieder regelmäßige Periodenblutungen und eine ungestörte Fertilität zu beobachten sind. Bei einer Kombinationstherapie mit Kortikosteroiden und Azathioprin sollte vor einer geplanten Schwangerschaft Azathioprin sicherheitshalber abgesetzt werden. Tritt jedoch die Gravidität unter einer derartigen Kombinationstherapie ein, sollte sie fortgeführt werden, da das Risiko für den Feten geringer einzuschätzen ist als das Risiko einer Exazerbation der AIH. Die Dosierung der Glukokortikoide ist wie vor der Schwangerschaft bei stabilem Krankheitsverlauf unverändert beizubehalten, bei entzündlichen Schüben sollte eine Dosissteigerung erfolgen. Eine derartige Therapie gilt für Mutter und Kind als relativ ungefährlich. Das gleiche gilt für Azathioprin in der vorgeschriebenen täglichen Dosis von 50-100 mg, auch wenn dieses Immunsuppressivum selten zur fetalen Wachstumsverzögerung und zur Knochenmarksdepression bei Mutter und Kind führen kann (16, 59, 77, 103). Es ist anzunehmen, dass bei der überwiegenden Zahl der Patientinnen eine Gravidität den Verlauf der AIH beeinflusst. Die Krankheitsaktivität kann gesteigert werden (16, 59, 172) oder – bisher deutlich häufiger – es kommt zur Remission ab dem 2. Trimenon, aber meist mit erneuter Verschlechterung postpartal (16, 26, 109, 172, eig. Beobachtungen) (Abb. 1.5 und 1.6). In Einzelfällen erfolgte der Nachweis der AIH erstmals im Wochenbett (59, 82, 133, 167). Wie bei der PBC mehren sich somit auch bei der AIH die Hinweise, dass die im Allgemeinen immunsuppressiv wirkende Schwangerschaft die Krankheitsaktivität ab dem 3. bis 4. Monat hemmt mit einem Rebound nach der Entbindung. Entsprechend kann in solchen Fällen unter engmaschiger Kontrolle die immunsuppressive Therapie reduziert werden (z. B. die Azathioprin-Dosis auf ca. 50 % der Ausgangsdosis), um postpartal die Ausgangsdosierung wieder aufzunehmen. Ein derartiger positiver Effekt ist auch bei einigen anderen Autoimmunkrankheiten zu beobachten, z. B. bei rheumatoider Arthritis, multipler Sklerose, Myasthenia gravis und bei chronisch entzündlichen Darmerkrankungen, nicht jedoch beim systemischen Lupus erythematodes. Als Erklärung wird angenommen, dass in der Schwangerschaft im Rahmen der physiologischen Immunsuppression die Dominanz der Th1- über Th2-Zellen abnimmt, so dass weniger, die Schwangerschaft gefährdende, proinflammatorische Zytokine gebildet werden. Östrogene, Progesteron, Androgene, Glukokortikoide, HCG und andere Faktoren unterstützen diese Immunsuppression (16, 59, 103).
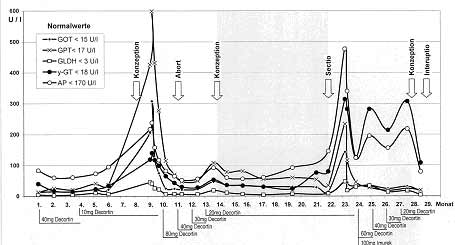
Abb. 1.5 AIH Typ III (SLA-positiv). Verlauf der Enzymaktivitäten im Serum bei einer 32-jährigen Patientin. 3 Konzeptionen, eine ausgetragene Schwangerschaft.
Abb. 1.6 AIH/PSC-Overlapsyndrom. Verlauf der Enzymaktivitäten im Serum während der Schwangerschaft und postpartal bei einer 33-jährigen Patientin (3-grav, 3-para). Bilirubin stets < 1,0 mg/dl. Keine medikamentöse Therapie. Somit können Frauen mit AIH bei stabiler Leberfunktion und normalem Bilirubinspiegel – ein entsprechendes Monitoring der Krankheitsaktivität vorausgesetzt – in etwa 80 % eine Schwangerschaft problemlos austragen. Insgesamt scheint aber die Abortrate, insbesondere bei schwerer verlaufender AIH, erhöht zu sein (14 – 24 %) (59, 172). Liegt allerdings eine Zirrhose vor, evtl. sogar mit Ösophagusvarizen und/oder Aszites, ist das mütterliche und fetale Morbiditäts- und Mortalitätsrisiko deutlich erhöht (Tab. 1.5). Über eine möglicherweise schwangerschaftsspezifische autoimmune Lebererkrankung in der Frühschwangerschaft in zwei Fällen berichtete 1993 eine japanische Arbeitsgruppe. In der 8. bzw. 10. Schwangerschaftswoche entwickelte sich eine akute Hepatitis. Histologisch fanden sich in enger Nachbarschaft zu den geschädigten Hepatozyten humanes Choriongonadotropin sowie entzündliche Infiltrate überwiegend aus CD8-Lymphozyten. HLA-DR-Antigen war auf den mononukleären Zellen nachweisbar. Ebenso lagen in der Dezidua lymphozytäre Infiltrationen vor. Nach der Schwangerschaft bildeten sich diese Alterationen spontan zurück. Die Autoren vermuten, dass die HCG-Ablagerungen in oder an den geschädigten Leberzellen das Zielantigen zytotoxischer T-Zellen darstellen (121). Die Frage, ob es sich hier um eine neue Form einer schwangerschaftsspezifischen Autoimmunhepatitis handelt, kann derzeit bei bisher erst zwei Beobachtungen nicht beantwortet werden (74). 1.2.7. Primär biliäre Zirrhose (PBC) und primär sklerosierende Cholangitis (PSC) Die PBC ist durch eine chronische nicht eitrige destruierende Cholangitis der kleinen intrahepatischen Gallengänge gekennzeichnet. Die Erkrankung tritt familiär gehäuft auf und betrifft zu 90 % Frauen mit einem Altersgipfel zwischen 40 und 59 Jahren. Die Ätiologie ist nicht bekannt, neben genetischen werden insbesondere immunologische Faktoren verantwortlich gemacht. Ebenfalls bislang nicht geklärt ist die hohe Dominanz des weiblichen Geschlechts bei der PBC. Als ein möglicher Auslöser werden die Geschlechtshormone diskutiert, da Östrogene und Gestagene Immunreaktionen anders kontrollieren als Androgene (103, 104, 133). Dagegen gilt ein Mikrochimärismus, das Einschwemmen embryonaler Zellen in den Kreislauf der Schwangeren, als Ursache für wenig wahrscheinlich (87). Charakteristisch für die PSC ist die chronische obliterierende Entzündung der intra- und/oder extrahepatischen Gallenwege. Die Manifestation liegt am häufigsten zwischen dem 25. und 40. Lebensjahr, zu 70 % sind Männer betroffen, in etwa 75 % besteht eine Assoziation zwischen PSC und einer chronisch entzündlichen Darmerkrankung. Auch hier wird neben anderen Faktoren ätiologisch ein Immunprozess in Betracht gezogen. Beide Erkrankungen führen mit variablem Verlauf zu progressiver Cholestase und schließlich zur biliären Zirrhose. Nach den bisher wenigen Beobachtungen von Schwangerschaften bei Patientinnen mit PBC kann man annehmen, daß die frühen Formen (Stadium I – III) durch eine Schwangerschaft nicht richtungsgebend verschlimmert werden. Ein unveränderter Verlauf wurde ebenso gesehen wie eine Zu- oder sogar Abnahme der Cholestase. In diesen Fällen war noch keine Behandlung mit Ursodeoxycholsäure (UDC) erfolgt. Aufgrund der positiven Daten bei ICP empfiehlt sich heute – und erste Mitteilungen belegen dies – die UDC-Therapie während der gesamten Schwangerschaft durchzuführen. Keine unerwünschten Wirkungen, sondern vielmehr positive Effekte auf die mütterliche Erkrankung und den Fetus werden gesehen. Etwa 80 % der Patientinnen können konzipieren, und gleichfalls in 80 % endet die Gravidität regelrecht. Die in einigen Fällen zu beobachtende Verbesserung der Leberfunktion während der Schwangerschaft, postpartal gefolgt von einem Anstieg der Krankheitsaktivität könnte wie bei der AIH auf eine Immunsuppression durch die foeto-plazentare Einheit zurückgeführt werden, vermittelt u. a. durch Östrogene und eventuell auch Gestagene (50, 70, 103, 104, 133, 142, 144, 149, 161). Die prognostische Einschätzung ist gänzlich anders beim Vorliegen eines kompletten zirrhotischen Umbaus (Stadium IV). Schwangerschaften in diesem Stadium sind eine ausgesprochene Rarität. Die Schwangere ist wie bei fortgeschrittenen Zirrhosen anderer Genese hochgradig gefährdet durch die Verschlechterung der Leberfunktion, durch Aszitesbildung und durch die Auslösung einer Ösophagusvarizenblutung. Entsprechend ist auch mit einer gesteigerten Früh- und Totgeburtlichkeit zu rechnen (Tab. 1.5). Unklar ist, ob in diesem späten Stadium die Gabe von UDC noch sinnvoll ist. Es liegt nahe, daß Schwangerschaften bei der PSC noch seltener sind (ca. 20 Fallmitteilungen) als bei der PBC. Auch hier sind im frühen Stadium komplikationslose Schwangerschaftsverläufe zu erwarten, dagegen bei der kompletten Zirrhose unter dem Einfluß der Gravidität Dekompensation und Varizenblutung sowie ein hohes Risiko für Frühgeburt und intrauterinen Fruchttod (Abb. 1.6). Die Datenbasis für die UDC-Therapie bei der PSC ist schmal. Gerade in der Schwangerschaft bei PSC mit der Gefahr der Akkumulation toxischer Gallensäuren sollte UDC jedoch verabreicht werden, da von einer protektiven Wirkung auf den Feten auszugehen ist (51, 85, 97, 103, 104). Anzumerken ist hier die fehlende Zulassung von UDC in der Schwangerschaft. Beim Auftreten fokaler Stenosen der großen Gallenwege sollten diese auch in der Schwangerschaft mittels endoskopischer Ballondilatation oder durch das Einsetzen von Stents therapiert werden. 1.2.8. Leberzirrhose Die Leberzirrhose ist als irreversibler Endzustand von chronisch progredienten Lebererkrankungen verschiedenster Ätiologie definiert. Sie ist durch Nekrosen, Entzündung, Regeneration und Bildung von Bindegewebssepten charakterisiert. Dieser chronische Umbau führt zu Funktionseinschränkungen und kann durch die Zerstörung des hepatischen Gefäßapparats zum Pfortaderhochdruck mit Ausbildung portosystemischer Kollateralen führen. Alkoholabusus und die Virushepatitiden B, C und D sind die häufigsten Ursachen einer Leberzirrhose in Westeuropa. Seltenere Ursachen sind autoimmune Erkrankungen, Stoffwechselerkrankungen und toxische Leberschäden (z. B. durch Medikamente). Laut Autopsiestudien muß von einer Prävalenz von ca. 9,5 % ausgegangen werden. Männer sind von der Entwicklung einer Leberzirrhose häufiger betroffen als Frauen. Schwangerschaften sind bei der Leberzirrhose unabhängig von den vielfältigen Ursachen selten (66, 69, 70, 74, 77, 149, 152, 163). Als Ursache der herabgesetzten Fertilität werden Amenorrhöen sowie anovulatorische Zyklen als Folge eines gestörten Sexualhormonstoffwechsels angesehen (64). Allerdings zeigt die zugrundeliegende hypothalamische-hypophysäre Dysfunktion keine strenge Korrelation zur Schwere der Lebererkrankung. Ein weiterer Grund für die erniedrigte Konzeptionsrate dürfte im höheren Lebensalter der zirrhosekranken Frau, zumindest in Westeuropa und in den USA, zu sehen sein. Das Durchschnittsalter der hier beobachteten Fälle betrug 30 Jahre, ein Alter, in dem die Konzeptionserwartung bereits herabgesetzt ist (Abb. 1.7) (69, 70). Eine verlässliche Aussage, welchen Einfluß eine Schwangerschaft auf den Verlauf der Leberzirrhose nimmt, läßt sich naturgemäß wie auch bei der chronischen Hepatitis nicht machen. Mit einer Aktivierung selbst eines beruhigten Prozesses muß jedoch – wie auch außerhalb der Gravidität – gerechnet werden. Gemessen an den Funktionsparametern wird in den meisten Fällen die chronisch entzündliche Lebererkrankung durch eine Schwangerschaft
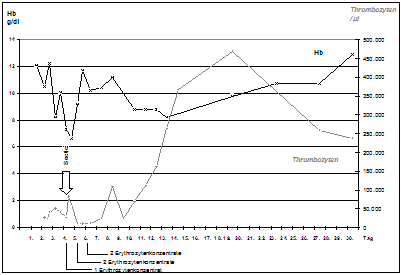
wenig beeinflußt. Die Ausgangslage, sei es ein aktives oder inaktives Stadium, wird meist beibehalten. Der häufig günstige Verlauf ist darauf zurückzuführen, dass die Konzeption überwiegend im kompensierten Stadium der Lebererkrankung erfolgt, die Schwangeren also eine positive Auslese darstellen. Im Wesentlichen wird die Prognose bei einer manifesten Leberzirrhose von dem Vorliegen einer portalen Hypertension und ihren Komplikationen (Aszites, Varizenblutung) bestimmt. Daß die Schwangerschaft in diesen Fällen zunehmend ein belastendes Moment darstellt, zeigt sich daran, daß bei 20 % aller Beobachtungen sich Aszites bildete und zwar überwiegend zum Schwangerschaftsende hin im dritten Trimenon und im Wochenbett (Abb. 1.8) (65, 69, 70). Pathogenetisch spielen in der Aszitesbildung Hypalbuminämie, Natriumretention und portale Hypertension die wesentliche Rolle, wobei diese Faktoren in der Schwangerschaft mit den bereits physiologischen Alterationen im Eiweißstoffwechsel und der vermehrten Produktion von Aldosteron sowie mit dem Anstieg des zirkulierenden Plasmavolumens und des Körperwassers noch eine Verstärkung erfahren. Weitere schwangerschaftsbedingte Veränderungen wie Kompressionswirkung des Uterus mit nachfolgender portaler Druckerhöhung sowie erhöhte Gefäßpermeabilität tragen gleichfalls zur Aszitesgenese bei. Welche Bedeutung der Erhöhung des Pfortaderdrucks für die Aszitesbildung zukommt, läßt sich allein aus der Tatsache ableiten, daß bei einem Drittel der bisher beobachteten Schwangerschaften mit Aszites Ösophagusvarizen nachgewiesen wurden. Die Prognose ist in
Abb. 1.8 Aszitesbildung in 33 Schwangerschaften mit Leberzirrhose (69, 70) diesem Falle deutlich eingeschränkt, fast die Hälfte der Patientinnen starb an einer Ösophagusvarizenblutung und/oder an einer Leberinsuffizienz (Tab. 1.5) (65, 68, 69, 70, 74, 77). In 45 % der bisher beschriebenen Fälle von Schwangerschaften bei Leberzirrhose bestanden Ösophagusvarizen als Folge eines intrahepatischen Blocks, die in 64 %, und zwar bevorzugt im zweiten und dritten Trimenon, bluteten mit einer mütterlichen Letalität von 13 %. Dieses vermehrte Auftreten von Ösophagusvarizenblutungen wird durch mehrere Faktoren begünstigt. Bereits in der normalen Schwangerschaft führen Hypervolämie und intraabdomineller Druckanstieg durch den wachsenden Uterus zu einer physiologischen portalen Hypertension. Durch die Kompresssion der V. cava inferior durch den Uterus erhöht sich der Blutabfluss über die V. azygos. Bei der Leberzirrhose pfropfen sich diese physiologischen Veränderungen den vorbestehenden pathogenetischen Faktoren, der bereits erhöhten Druck- und Volumenbelastung im Pfortadersystem, auf (Abb. 1.9) (68, 69, 70, 74, 88, 107, 162).
Abb. 1.9 Häufigkeit von Ösophagusvarizenblutungen bei 29 Schwangeren mit prähepatischem Block und bei 34 Schwangeren mit intrahepatischem Block bei Leberzirrhose (69, 70). Chirurgische Therapieverfahren beim Pfortaderhochdruck werden heute, da sie technisch aufwendig und relativ risikoreich sind, nur noch selten eingesetzt. Wie frühere Untersuchungen zeigen, vermögen vor allem porto-systemische Shuntoperationen auch in der Schwangerschaft effektiv den portalen Druck zu senken. Shuntoperationen, deren Durchführung sogar noch in den ersten beiden Trimestern möglich ist, senkten die Blutungsfrequenz auf 7 % mit einer mütterlichen Letalität von 2 %. Als die wirksamste Behandlungsmaßnahme bei akuter Varizenblutung werden heute auch in der Schwangerschaft die Ligatur und/oder die Sklerotherapie angesehen. Als Alternative bietet sich auch der transjuguläre Stentshunt (TIPS) an. Bei bekannter Leberzirrhose empfiehlt es sich, vor und während der Schwangerschaft Ösophagusvarizen prophylaktisch durch Injektion von Polidocanol oder durch Varizenligatur zu behandeln. Der endoskopischen Varizenligatur sollte heute der Vorzug gegeben werden, da über mögliche toxische Nebenwirkungen des Polidocanols an der Frucht wenig bekannt ist. Anzustreben ist, Ösophagusvarizen bereits vor Eintritt einer Konzeption prophylaktisch zu sklerosieren. Vor oder adjuvant zur Ligatur- oder Sklerosierungstherapie bei blutenden Varizen kann auch in der Schwangerschaft eine medikamentöse Therapie mit ß-Blockern (Propanolol) oder mit Octreotid, dem länger wirksamen Analog des Somatostatins, erfolgen. Ungeeignet sind Vasopressin-Analoga, die bei Mutter und Kind zu verschiedenen vasospastischen Komplikationen sowie zur Plazentaablösung führen können. Systematische Untersuchungen zur Effektivität einer derartigen endoskopischen und medikamentösen Therapie in der Schwangerschaft fehlen allerdings bisher. Auf eine länger dauernde Ballontamponade (mehr als 24 Stunden) sollte wie außerhalb der Schwangerschaft wenn irgend möglich verzichtet werden, da die sich entwickelnden Wandläsionen deutlich das Komplikationsrisiko der endoskopischen Therapie erhöhen (69, 70). Zur Vermeidung der Aszitesbildung und zur Senkung des Plasmavolumens und somit auch zur Varizenblutungsprophylaxe empfehlen sich die Kochsalzrestriktion in der Nahrung und gegebenenfalls die behutsame diuretische Therapie. Beim Vorliegen eines Aszites ist ebenso vor einer forcierten medikamentösen Diurese zu warnen wie vor therapeutischen Aszitespunktionen. Eine Entlastung des Aszites ist nur unter der Geburt bei Eintritt von Wehenruhe zu vertreten, da in diesem Falle Bauchmuskulatur und Zwerchfell – durch den Aszites überdehnt – in ihrer Kontraktionsfähigkeit weitgehend eingeschränkt sind. Bei portaler Hypertension sollte zur Vermeidung einer weiteren Druckerhöhung die Austreibungsperiode durch assistierte operative Entbindung (Vakuumextraktion, Forzeps) beendet werden. Besteht eine dekompensierte Leberzirrhose mit portaler Hypertension, kann im Einzelfall eine Abruptio durchaus erwogen werden. Die Schwangere ist nicht nur durch die Leberinsuffizienz, sondern auch durch die Auslösung einer Varizenblutung sowie durch einen Aszites und dessen forcierter Therapie hochgradig gefährdet. Dieses Risiko wird durch die Abruptio vermindert, da der Eingriff die hämodynamische Situation unmittelbar entlastet. Aus diesen Ausführungen folgt, daß nicht die Leberinsuffizienz durch die Belastung des Organs in der Schwangerschaft die Hauptgefahr für die Leberzirrhotikerin darstellt, sondern die portale Hypertension. Ösophagusvarizenblutung und Ausbildung eines Aszites bestimmen weitgehend die Prognose und bedingen eine mütterliche Sterblichkeit von durchschnittlich 8 %. Auch die kindliche Prognose erfährt bei einer Leberzirrhose der Mutter mit einer erhöhten Zahl von Frühgeburten und perinatal Verstorbenen eine Einschränkung. Es zeigt sich, daß die Frühgeburten- und Totgeburtenrate umso mehr ansteigt, je florider bzw. fortgeschrittener der entzündliche Prozeß ist, also besonders bei ikterischen Schüben. Besonders verschlechtert wird die Prognose des Kindes ebenfalls bei Ösophagusvarizenblutungen und Vorliegen von Aszites. Varizenblutungen führten in 32 % der Fälle zum Tode des Kindes und in 27 % zur Frühgeburt, bei Bestehen eines Aszites starben 40 % und 28 % der Fälle waren Frühgeburten (Tab. 1.5 und 1.8) (69, 70). Erforderlich ist auch bei der Leberzirrhose die Bestimmung der virologischen Parameter, um bei einer HBV-Infektion der Mutter die Infektion des Kindes durch eine passiv-aktive Simultanprophylaxe direkt nach der Geburt zu verhindern (70, 74, 78). Tab. 1.8 Kontraindikationen für eine Schwangerschaft und Indikationen zur Abruptio bzw. vorzeitigen Entbindung
1.2.9. Portale Hypertension Für die Entstehung des Pfortaderhochdrucks sind pathogenetisch zwei Komponenten von Bedeutung: Zum einen eine Erhöhung des Strömungswiderstandes im portalen Stromgebiet (durch mechanische Kompression oder Vasokonstriktion) und zum anderen durch eine Zunahme des splanchnischen und portalen Blutflusses. Es besteht eine komplexe hyperdyname Kreislaufsituation mit den klinischen Zeichen des erhöhten Herzminutenvolumens, der Tachykardie und der arteriellen Hypotonie. Die portale Druckerhöhung führt zur verminderten Leberdurchblutung und zur Bildung von Kollateralen zwischen Pfortader und Vena cava. Als Folgen können sich klinisch eine Leberfunktionsverschlechterung oder Komplikationen wie Ösophagusvarizenblutung, Aszites und Enzephalopathie einstellen. Die Einteilung der portalen Hypertension erfolgt in der Regel nach der anatomischen Lokalisation der Widerstandserhöhung. Bei der prähepatischen Widerstandserhöhung (z. B. durch Pfortader- oder Milzvenenthrombose, arterioportale Fisteln) ist die Leberfunktion nicht oder nur gering eingeschränkt und auch ein Aszites ist sehr selten. Bei der intrahepatischen portalen Hypertonie wird eine präsinusoidale (z. B. Schistosomiasis), eine sinusoidale (z.B. Leberzirrhose, Fettleber) und eine postsinusoidale Form (z.B. Budd-Chiari-Syndrom) unterschieden. Ursachen des posthepatischen Pfortaderblocks sind Druckerhöhungen im rechten Herzvorhof (Pericarditis constrictiva, Rechtsherzinsuffizienz) bzw. Thrombose, Membranen oder Tumorinvasion der V. cava inferior. Die Aszitesbildung ist im Wesentlichen auf den erhöhten Druck in den Sinusoiden bei der sinusoidalen und postsinusoidalen Lokalisation zurückzuführen. In den westlichen Industrieländern sind bei Erwachsenen die alkoholischen und viralen Leberzirrhosen die häufigsten Ursachen der portalen Hypertonie, bei Kindern ist die Pfortaderthrombose ätiologisch führend. Eine Splenomegalie kann bei allen Formen des Pfortaderhochdrucks gefunden werden. Wie beim intrahepatischen Block der Leberzirrhose sind auch Schwangerschaften beim prähepatischen Block wie bei der intrahepatischen präsinusoidalen Hypertension selten (2, 18, 32, 67, 68, 69, 70, 74, 107, 149, 152, 163, 193). Der Grund hierfür ist nicht bekannt, sind doch Leberfunktion und Sexualhormonstoffwechsel bei diesen Blockformen nicht gestört. Die Ausbildung von Ösophagusvarizen stellt auch hier die wichtigste Komplikation dar. Das Durchschnittsalter der bisher beobachteten Frauen mit prähepatischem Block beträgt 24 Jahre und liegt damit um 6 Jahre unter dem Durchschnittsalter der Frauen mit intrahepatischem sinusoidalen Block bei Leberzirrhose (Abb. 1.7). Bei allen Patientinnen war der Block bereits vor der Schwangerschaft ausgebildet und bei den meisten hatten bereits im Kindesalter schwere Intestinalblutungen eine chirurgische Intervention in Form von Palliativeingriffen und Shuntoperationen erforderlich gemacht. Trotz dieser chirurgischen Eingriffe kam es während der Schwangerschaft in 40 % zu Ösophagusvarizenblutungen und zwar in etwa dem gleichen Prozentsatz nach Shuntoperationen wie nach alleinigen Palliativmaßnahmen. Dies ist darauf zurückzuführen, daß Shunts, die im Kindes- und Jugendalter angelegt wurden, meistens ineffektiv sind so wie auch Palliativoperationen keinen dauernden drucksenkenden Effekt haben. Es sind die gleichen Faktoren wie bei der Leberzirrhose, die hier in der Schwangerschaft eine Varizenblutung auslösen und folglich ist es mit dem zweiten und dritten Trimenon der Zeitraum, in dem die Varizenblutung gehäuft auftritt (Abb. 1.9) (67, 68, 69, 70). Mit einer mütterlichen Letalität von 3 % und einer erhöhten Zahl von Totgeburten erfährt die mütterliche und kindliche Prognose ebenfalls eine Einschränkung, allerdings nicht in dem Maße wie bei Varizenblutungen infolge Leberzirrhose. Wie bei der Leberzirrhose mit portaler Hypertension gelten hier die gleichen therapeutischen Überlegungen. Bei weitgehend kompensiertem prähepatischen Pfortaderhochdruck, d.h. bei Ausbildung von effektiven spontanen oder operativen Anastomosen, ist am ehesten ein komplikationsloser Schwangerschaftsverlauf zu erwarten. Beim Nachweis von Ösophagusvarizen sollte bereits vor der Schwangerschaft eine prophylaktische Varizeneradikation durch Gummibandligatur oder Sklerosierung erfolgen (Tab. 1.5 und 1.8) (69, 70, 74). Ein partieller oder vollständiger thrombotisch bedingter Verschluss der Lebervenen (Budd-Chiari-Syndrom) ist eine seltene Schwangerschaftskomplikation, die sich weniger in der Spätschwangerschaft als vielmehr im Wochenbett und in der Beobachtungszeit bis zu 9 Monaten post partum manifestiert. Zahlreiche Ursachen werden als pathogenetische Faktoren diskutiert, in Schwangerschaft und postpartaler Phase liegen wahrscheinlich mehrere thrombogene Faktoren vor wie erhöhte Östrogen- und Progesteron-Spiegel, angeborene und erworbene Gerinnungsstörungen, Blutvolumenvermehrung und Strömungsverlangsamung im Abdominalbereich. Das Vorliegen einer aktivierten Protein-C-Resistenz als Folge einer Mutation von Faktor V ist wahrscheinlich der wichtigste Risikofaktor für das Auftreten einer derartigen venösen Thrombose. Das klinische Bild kann über Wochen bis Monate schleichend beginnen oder akut einsetzen. Bei der häufigeren chronischen Verlaufsform finden sich als führende klinische Symptome Aszites, Hepatosplenomegalie, Ösophagusvarizen, Ikterus und obere Intestinalblutung. Auch wenn bisher nur wenige Fälle eines Budd-Chiari-Syndroms im Zusammenhang mit einer Schwangerschaft beobachtet wurden, lassen sich dennoch hieraus einige Schlüsse ziehen: Besteht bei einer jungen Frau der Verdacht auf ein Budd-Chiari-Syndrom, muß mit allen Mitteln die Diagnose gestellt oder ausgeschlossen werden (Farbdoppler-Sonographie). Finden sich Hinweise auf eine solche Erkrankung, ist eine Schwangerschaft absolut kontraindiziert, ebenso eine orale Kontrazeption. Tritt die Thrombosierung der Lebervenen während der Schwangerschaft auf, besteht nach derzeitiger Auffassung die Indikation zum Abbruch der Schwangerschaft, da die Erkrankung durch die Schwangerschaft richtunggebend verschlimmert wird (Tab. 1.5 und 1.8). Umgehend zu diskutieren ist heute jedoch auch eine Lebertransplantation höchster Dringlichkeitsstufe in der Schwangerschaft (42, 54, 68, 69, 70, 74, 152). 1.2.10. Lebertransplantation Schwangerschaften nach orthotoper Lebertransplantation sind, nachdem 1978 die erste erfolgreiche Gravidität nach Lebertransplantation beschrieben wurde, insgesamt noch selten. Schwangerschaft nach Leber-, Nieren- oder Herztransplantation gelten als Risikoschwangerschaft für Mutter und Fetus und machen eine intensive interdisziplinäre Betreuung notwendig. Die Risiken für die Mutter bestehen in einer erhöhten Inzidenz an EPH-Gestosen, vermehrten bakteriellen, viralen und fungalen Infekten wie auch in Abstoßungsreaktionen. Dem Feten drohen Frühgeburt, intrauterine Wachstumsretardierung, pränatale Infektionen sowie Fehlbildungen. Zur immunsuppressiven Therapie bei Schwangerschaften nach Lebertransplantation stehen Kortikosteroide, Cyklosporin, Tacrolimus und Azathioprin zur Verfügung. Von besonderer Bedeutung sind dabei neben der Funktion der transplantierten Leber die möglichen Nebenwirkungen dieser Immunsuppressiva sowie die fetale Entwicklung. Auch wenn nach den bisherigen beschränkten Erfahrungen die Teratogenität dieser Medikamente als nicht erhöht anzunehmen ist, so ist es im Einzelfall doch nicht möglich, speziell bei verschiedenen Kombinationen, eine sichere prognostische Aussage zu treffen (5, 32, 78, 143, 152, 199). 1.2.11. Lebertumoren Primäre Lebertumoren umfassen ein großes Spektrum von benignen und malignen epithelialen wie nichtepithelialen Tumoren, aber auch von tumorartigen Läsionen. Obwohl im gebärfähigen Alter selten, sollte die Detektion und Differenzierung dieser umschriebenen Leberveränderungen bereits vor Eintritt einer Schwangerschaft erfolgt sein. Wegen des Rupturrisikos in der Schwangerschaft empfiehlt es sich, bei großen (7 – 10 cm), oberflächlichen und resezierbaren Läsionen die chirurgische Intervention zu erwägen. Dies trifft insbesondere beim Vorliegen eines hepatozellulären Adenoms zu. Gerade in der Schwangerschaft, in der den Östrogenen (wie auch bei Einnahme von Kontrazeptiva) gefäßerweiternde und proliferative Effekte zugeschrieben werden, können intraabdomineller Druckanstieg und erhöhte Bauchdeckenspannung (z. B. beim Pressen oder Erbrechen) oder die Volumenvermehrung im prähepatischen Raum unter der Geburt eine Ruptur auslösen. Auf die spontane Leberruptur, die im Rahmen eines HELLP-Syndroms auftritt, wird im Kap. 1.3.2. eingegangen. Diagnostisch ist bei typischen Befunden die sonographische Charakterisierung ausreichend (B-Bild- und Farbdoppler-Sonographie, Einsatz von Ultraschallkontrastmitteln). Bei Zweifeln werden CT, MRT und letztlich die histologische Untersuchung eingesetzt. Selten bedarf es der histopathologischen Aufarbeitung der gesamten resezierten Läsion, um eindeutige Diagnosen zu erhalten. Kongenitale Leberzysten gehören mit einer Prävalenz von 3 – 5 % zu den häufigen umschriebenen Läsionen der Leber, die bei Frauen fast doppelt so häufig wie bei Männern angetroffen werden. Bei Größenzunahme können Zysten Druck- und Völlegefühl wie Schmerzen hervorrufen, Einblutung und Infektion sind seltene Komplikationen (169, 170). In der Schwangerschaft führen kongenitale Zysten in der Regel nicht zu Beschwerden. Sollte es jedoch zu starkem Druck- und Völlegefühl oder zu Verdrängungserscheinungen an den Nachbarorganen kommen, kann auch in der Schwangerschaft die gezielte perkutane Punktion vorübergehend Entlastung bringen. Die Peliosis hepatis ist überwiegend durch bis zu 1 cm, selten bis zu 3 cm große, mit Endothel ausgekleidete blutgefüllte Räume, die mit den Sinusoiden in Verbindung stehen, charakterisiert. Das Befallsmuster ist fokal oder diffus. Ätiologisch wird die Peliosis hepatis im Zusammenhang mit der Gabe von oralen Kontrazeptiva sowie von anabolen und androgenen Steroiden gebracht. Über die Blutungs- oder Rupturneigung von Peliosisherden während der Schwangerschaft gibt es keine Angaben. Einzelfallberichte über spontane Leberrupturen bei sonst gesunden Patienten lassen es aber angebracht erscheinen, in der Schwangerschaft bei ausgeprägter Peliosis und kapselnahen Herden sowie beim Auftreten einer schmerzhaften Hepatomegalie die Leber engmaschig mit bildgebenden Verfahren zu kontrollieren. Das kavernöse Hämangiom ist mit einer Inzidenz zwischen 0,4 und 20 % in Sektionsstatistiken der häufigste benigne Lebertumor. Er lässt sich in allen Altersgruppen nachweisen, am häufigsten allerdings bei Erwachsenen, und zwar bei Frauen mittleren Alters. Entsprechend tritt diese Läsion bei Frauen zwei- bis sechsmal häufiger auf als bei Männern. Die Beobachtungen, dass dieser Tumor bei Frauen nicht nur häufiger, sondern meistens auch größer ist, dass er bei Mehrgebärenden häufiger ist und dass er während Pubertät und Schwangerschaft oder unter oraler Antikonzeption an Größe zunehmen kann, lässt eine wachstumsfördernde Rolle der Geschlechtshormone vermuten. Über drei Viertel der Hämangione sind solitär und sowohl subkapsulär als auch innerhalb beider Leberlappen gelegen. Sie sind meist klein mit einer Größe von wenigen Millimetern bis zu etwa 4 cm. Riesenhämangiome über 5 cm finden sich bei unter 10 % der Patienten. Histologisch liegen mit Endothel ausgekleidete kommunizierende vaskuläre Hohlräume vor, die durch Bindegewebssepten getrennt und durch eine fibrosierte Kompressionszone vom umgebenden Leberparenchym abgegrenzt sind. Die kleineren Hämangiome sind in der Regel asymptomatisch, größere können zu Oberbauchbeschwerden führen. Akute Schmerzen treten bei Blutungen und bei Thrombosen im Hämangiom auf sowie – sehr selten – bei Tumorruptur mit Ausbildung eines Hämoperitoneums. In der Schwangerschaft sind Tumorgrößen über 5 cm, eine oberflächliche Lage, eine Größenzunahme während der Schwangerschaft sowie akut einsetzende Bauchschmerzen als Risikofaktoren für eine Ruptur anzusehen, die entsprechend zu kontrollieren sind (24, 45, 52, 56, 81, 111, 165, 170). Die fokal noduläre Hyperplasie (FNH) ist nach den Hämangiomen der zweithäufigste benigne Lebertumor. Wahrscheinlich entsteht die FNH als Folge von Gefäßmissbildungen. Die Prävalenz beträgt etwa 3 %. Frauen zwischen dem 22. und 50. Lebensjahr sind 6- bis 8-mal häufiger als Männer betroffen. Aufgrund früherer klinisch-epidemiologischer Studien wurde angenommen, dass das Tumorwachstum ähnlich wie beim Adenom und Hämangiom hormoninduziert sei. Ein Zusammenhang wurde mit den endogenen Östrogenen, der langjährigen Einnahme von oralen Kontrazeptiva, dem erhöhten Blutungsrisiko unter diesen Hormonen und der möglichen Rückbildung der FNH nach Absetzen der Kontrazeptiva gesehen. Für die Schwangerschaft wurde ein erhöhtes Risiko für Wachstum und Ruptur einer bestehenden FNH angenommen. Neuere Daten weisen jedoch daraufhin, dass weder orale Kontrazeptiva Größe oder Zahl der FNH-Läsionen beeinflussen noch die Schwangerschaft Veränderungen oder Komplikationen hervorruft (19, 24, 112, 170, 195). In der Regel findet sich eine solitäre FNH meist kleiner als 5 cm (1 – 20 cm), in etwa 20 % liegen mehrere Knoten vor. Häufig ist die FNH subkapsulär lokalisiert, jedoch auch in beiden Leberlappen oder selten als gestielter Tumor. Eine Größenzunahme der FNH (über 7 cm) führt zu Hepatomegalie und Druckschmerz. In etwa 10 % der Fälle sind als Komplikationen Einblutungen, Nekrosen oder Ruptur zu fürchten. Aus diesem Grunde halten wir, auch wenn die meist kleinere FNH asymptomatisch bleibt, in der Schwangerschaft eine Verlaufskontrolle mittels Sonographie für angezeigt. Das hepatozelluläre Adenom ist seltener als die FNH, in Sektionen findet sich eine Prävalenz unter 0,5 %. Vor der Ära der Kontrazeptiva waren Adenome Raritäten. Nachdem Baum et al. (11) 1973 erstmals auf einen Kausalzusammenhang zwischen Langzeitanwendung von Antikonzeptiva und Leberzelladenomen hingewiesen hatte, erhöhte sich die Zahl der Beobachtungen rasch (113). Entsprechend findet sich das Adenom fast nur bei Frauen im gebärfähigen Alter (ca. 90 %) meist mit einer Antikonzeptiva-Einnahme von mehr als 5 Jahren. Die Inzidenz nimmt mit der Dauer der Einnahme, der Hormonmenge der Präparate und dem Alter der Frauen (über 30 Jahre) zu. Heute dürfte die Zahl der Adenome bei den modernen Antikonzeptiva mit niedrigen Hormonkonzentrationen wieder rückläufig sein. Adenome sind meistens solitär und überwiegend im rechten Leberlappen in einer sonst normalen Leber lokalisiert. Die Größe variiert zwischen 2 und 30 cm. Gewöhnlich entwickeln sich die Adenome oberflächennah unter der Kapsel. Multiple Adenome mit bis zu über 10 Herden sind selten und stellen möglicherweise ein eigenständiges Krankheitsbild (Leberadenomatose) dar, sie werden aber auch nach Langzeiteinnahme von Anabolika und im Rahmen der Glykogenspeicherkrankheiten Typ Ia und seltener Typ III beobachtet. In 2 eigenen, oben beschriebenen Beobachtungen von Glykogenose Typ Ia besteht eine Leberadenomatose (s. Kap. 1.2.3.4.). Das Überwiegen von Leberzelladenomen bei Frauen und die Assoziation mit der Langzeiteinnahme von Kontrazeptiva lässt einen hormonellen Einfluss auf die Pathogenese annehmen. Die wesentliche Rolle dürfte den Östrogenen zukommen, allerdings sind die genauen Pathomechanismen nicht bekannt. Östrogene stimulieren die Zellproliferation und fördern die Neubildung von Gefäßen und von peliosisartigen Arealen. Darüber hinaus induzieren Östrogene Gefäßerweiterungen, Verdickungen der Intima und Hyperplasien der Media von Venen und Arterien. Einerseits bilden sich Adenome in Einzelfällen nach Absetzen der Kontrazeptiva zurück, andererseits können sich während der Schwangerschaft unter der erhöhten hormonellen Stimulation die Läsionen aufgrund der vermehrten Vaskularisation vergrößern. Dadurch ist die Rupturgefahr, auch im Wochenbett, erhöht. Beim Nachweis eines Adenoms sind Kontrazeptiva und Anabolika abzusetzen. Bei kleinen Adenomen, zunächst asymptomatisch und meist Zufallbefunde, ist die sorgfältige sonographische Verlaufskontrolle vorab ausreichend. Symptomatisch werden Adenome bei Größenzunahme, infolge Einblutung und Nekrose sowie bei Ruptur der Läsion. Es empfiehlt sich daher bei über 5 cm großen resezierbaren Läsionen die operative Intervention. Die grundsätzliche Indikation zur Resektion des Adenoms ist jedoch wegen der erhöhten Komplikationsgefahr gerade gegen Ende der Schwangerschaft oder nach der Entbindung bei Frauen mit Kinderwunsch gegeben. Wegen der oft ausgeprägten Vaskularisation und der fehlenden Bindegewebskapsel ist die Blutungs- und Rupturgefahr beim Adenom größer als bei der bindegewebsreichen FNH (24, 60, 159, 170, 178). In Einzelfällen ist bei östrogenbehandelten Patientinnen sowie im Gefolge endokrin aktiver Tumoren das gleichzeitige Auftreten von Leberzelladenom und FNH dokumentiert. Eine eigene Beobachtung einer 33-jährigen Patientin wies 12 Leberrundherde mit echoarmem Randsaum, geringer venöser Randvaskularisation und zum übrigen Lebergewebe isoechogener Binnenstruktur auf. Der mit 6 x 3 x 3 cm größte Rundherd, in einigen Bereichen sehr inhomogen, fand sich im 6. Segment. Klinisch bestand eine ungewohnte Mattigkeit, klinisch-chemisch waren die Aktivitäten von AP und γ-GT erhöht. Vorausgegangen war eine orale Kontrazeption über 16 Jahre mit Ethinylestradiol in der Kombination mit Desogestrel, Norgestimat oder Levonorgestrel. Mit bildgebenden Verfahren war eine sichere Diagnose nicht möglich. Die sonographisch gezielte Punktion ergab normales Lebergewebe, und erst die Laparoskopie mit Biopsie führte zur Diagnose von multifokalen Leberzelladenomen. Bei sonographisch fehlender Tumorregredienz nach 12 Monaten unter Hormonkarenz willigte die Patientin schließlich in die Resektion des größten suspekten Herdes im Segment 6 sowie eines weiteren Herdes ein. Erst jetzt war mit dem seltenen Befund einer hyperplastisch-adenomatösen FNH, d.h. eines Mischtyps aus Adenom und FNH eine eindeutige histopathologische Zuordnung zu treffen (93). Das hepatozelluläre Karzinom (HCC) ist der häufigste primäre maligne Lebertumor. Die Inzidenz zeigt eine sehr unterschiedliche geographische Verteilung. In Westeuropa und den USA als Gebieten mit geringer Inzidenz liegt die Zahl der Neuerkrankungen für Männer bei 3 – 4, für Frauen bei 1 – 2 pro 100.000 Einwohner pro Jahr. Verschiedene Risikofaktoren, einzeln oder kombiniert einwirkend, werden diskutiert. Hierzu zählen u.a. chronische Virushepatitiden (HBV, HCV, HDV), Toxine (Alkohol, Aflatoxin etc.) und genetische Faktoren (Hämochromatose, Tyronsinämie, hepatische Porphyrien etc.), aber auch Kontrazeptiva und Anabolika werden als wahrscheinliche Risikofaktoren angesehen. In 60 – 90 % der Fälle ist das HCC mit einer Leberzirrhose assoziiert. In Ländern mit geringer Inzidenz liegt das mittlere Manifestationsalter zwischen dem 50. und 60. Lebensjahr. In den afrikanischen und asiatischen Hochinzidenzgebieten findet sich dagegen der Erkrankungsgipfel zwischen dem 20. und 35. Lebensjahr. Dies weist auf die besondere Bedeutung des Hepatitis B-Virus als Karzinogen hin, wobei die Infektion durch vertikale perinatale oder horizontale Transmission während der Kindheit übertragen wird. In den letzten Jahren nimmt die HCC-Inzidenz, auch bei jüngeren Menschen in den Industrienationen zu, was im Wesentlichen auf chronische Hepatitis C-Infektionen zurückgeführt wird. Eine Variante des HCC stellt das fibrolamelläre Karzinom dar (ca. 20 % der Fälle), das vor allem bei jüngeren Frauen in einer nichtzirrhotischen Leber auftritt mit besserer Prognose als das HCC (169). Männer mit Zirrhose erkranken zwei- bis viermal häufiger an einem HCC als Frauen, außerdem tritt der Tumor bei Männern früher auf. Die Gründe hierfür sind noch weitgehend unklar. Es liegt nahe, auch eine Beteiligung von Sexualhormonen in der Pathogenese des HCC anzunehmen. Epidemiologische, klinische und experimentelle Studien zeigen, dass regenerierende und proliferierende Prozesse in der Leber durch Östrogene stimuliert werden. Tierexperimentell erwiesen sich Östrogene als Induktoren und Promotoren von Lebertumoren, in epidemiologischen Untersuchungen wird die Langzeiteinnahme oraler Kontrazeptiva bei Frauen mit einer erhöhten Prävalenz einer FNH und einer HCC in Zusammenhang gebracht (129). Das HCC bei Männern exprimiert vermehrt Östrogenrezeptoren mit unterschiedlichem Rezeptorstatus. Neben dem Wildtyp existieren jedoch überwiegend mutierte Transkripte, die nicht mehr in der Lage sind, Östrogene zu binden (191). Dennoch könnten die bei Zirrhotikern erhöhten Östrogenkonzentrationen die Proliferation der Tumorzellen verstärken. Allerdings führte eine antiöstrogene Therapie mit Tamoxifen zu keiner Überlebensverlängerung (10).Weiterhin finden sich im HCC Rezeptoren für Androgene in höheren Konzentrationen, so dass auch Androgene in der Tumorentwicklung von Bedeutung sein könnten (127, 169). Anders stellt sich die Situation offensichtlich bei Frauen dar. Hier kommt möglicherweise im Erwachsenenalter den Östrogenen ein protektiver Effekt auf die Entwicklung eines HCC zu. In einer multizentrischen Fall-Kontrollstudie werteten Yu et al. (201) die Auswirkungen reproduktiver Faktoren auf das HCC-Risiko aus und prüften gleichzeitig, ob sich die Assoziation zwischen diesen Faktoren und dem HCC bei HBV-infizierten und HBV-negativen Frauen unterscheidet. 218 Frauen mit HCC sowie 729 weibliche Kontrollen, überwiegend Verwandte ersten Grades der Erkrankten, wurden in die Studie aufgenommen. Das Risiko für die Bildung eines HCC war umgekehrt proportional zu der Zahl normal verlaufender Schwangerschaften und dem natürlichen Menopausenalter der Frauen (45 – 55 Jahre). Das vorzeitige Sistieren der Ovarialfunktion (z.B. Ovarektomie) war ein Risikofaktor. Eine zeitlich begrenzte Hormonsubstitutionstherapie war im Gegensatz zur längerfristigen Therapie mit einem niedrigeren Risiko assoziiert. Eine HBV-Infektion hatte keinen Einfluss auf die reproduktiven Faktoren. Lediglich eine frühzeitig einsetzende Menarche (unter 12 Jahre im Vergleich zu über 16 Jahre) erhöhte das HCC-Risiko bei HBV-Infizierten gegenüber den HBV-negativen Frauen. Diese Daten erklären zum Teil, dass auch in der Schwangerschaft das HCC eine ausgesprochene Rarität darstellt. In den letzten 50 Jahren wurden weltweit annähernd 50 Fälle beschrieben. Nicht nur das seltenere und spätere Auftreten des HCC bei Frauen im Vergleich zu den Männern, sondern auch eine reduzierte Fertilität bei Vorliegen einer Leberzirrhose lassen sich zur Erklärung anführen. Da in Hochinzidenzgebieten relativ häufiger jüngere Menschen erkranken, stammen auch die meisten Beobachtungen aus diesen Regionen. Die überwiegende Zahl der Patientinnen wies eine Leberzirrhose oder eine HBV-Infektion auf, teils sind die Angaben zu Risikofaktoren unvollständig oder fehlen ganz. Wenige Frauen wiesen weder eine Leberzirrhose noch eine HBV- oder HCV-Infektion auf. In 3 Fällen lag ein fibrolamelläres Karzinom vor. Wie beim Leberzelladenom ist in der Schwangerschaft in gleicher Weise für das HCC eine erhöhte Rupturgefahr gegeben. Die medianen Überlebenszeiten von Schwangeren mit HCC sind kürzer als bei nichtschwangeren Frauen. Günstiger dagegen ist die Prognose bei Vorliegen eines fibrolamellären Karzinoms. Es ist zu fragen, ob bei der Frau der oben beschriebene „protektive“ Effekt der Östrogene beim manifesten HCC sistiert und die Steroidhormone insbesondere in der Schwangerschaft vielmehr jetzt zu einer Beschleunigung der Wachstumskinetik beitragen. In der Schwangerschaft unterscheidet sich die Symptomatik des HCC in fortgeschrittenen Stadien nicht von der bei nichtschwangeren Patientinnen (20). Die Diagnostik erfolgt durch bildgebende Verfahren einschließlich gezielter Punktion. Problematisch ist die Bestimmung der a-Fetoproteinkonzentration (AFP) im Serum als Tumormarker. Nur etwa in 50 – 90 % der Patienten mit HCC findet sich eine AFP-Erhöhung, AFP ist nicht spezifisch für ein HCC und Sensitivität und Spezifität sind mäßig. Konzentrationen über 500 ng/ml werden als nahezu beweisend für ein HCC angesehen. Werte über 200 ng/ml können aber auch physiologischerweise in der normalen Schwangerschaft erreicht werden. AFP wird ab dem ersten Trimenon in der fetalen Leber produziert und ist ab diesem Zeitpunkt im fetalen Kreislauf, Fruchtwasser und maternalem Serum nachweisbar. Höhere AFP-Konzentrationen finden sich auch bei einer Vielzahl angeborener Fehlbildungen, insbesondere Neuralrohr- und Bauchwanddefekten. Ein erhöhtes AFP in der 16. Schwangerschaftswoche kann also durchaus ein Hinweis auf Missbildungen sein, die durch gezielte Sonographie ausgeschlossen werden müssen. Bei unklarem sonographischen Befund empfiehlt sich die simultane Bestimmung des AFP im maternalen Serum und im Fruchtwasser. Beim Nachweis eines HCC in der Schwangerschaft stellt sich, wie außerhalb der Schwangerschaft, als erstes die Frage, ob eine Option für die derzeit einzigen potenziell kurativen Therapien wie Leberteilresektion, Lokalablation kleiner Tumorherde und Lebertransplantation besteht. Diese operativen Verfahren dürften auch hier nur für etwa ein Fünftel der Fälle in Frage kommen. In den beschriebenen Fällen wurden entsprechend unterschiedliche Therapieverfahren eingesetzt. Letztlich wird in interdisziplinärer Zusammenarbeit im Wesentlichen abhängig vom genauen Schwangerschaftsalter, dem Tumorstadium und dem Zustand der tumorfreien Leber im Einzelfall zu klären sein, welche Therapieformen in Betracht zu ziehen sind. Ebenso sollte die Indikation zum Schwangerschaftsabbruch und die Art des geburtshilflichen Vorgehens multidisziplinär getroffen werden (4, 24, 80, 84). 1.3 Schwangerschaftsspezifische Lebererkrankungen 1.3.1. Intrahepatische Schwangerschaftscholestase (ICP) Die ICP (idiopathischer Schwangerschaftsikterus) bildet unter den schwangerschaftsspezifischen Lebererkrankungen die größte Gruppe, die Erkrankungshäufigkeit ist in Deutschland vergleichbar mit den Erkrankungszahlen bei der akuten Virushepatitis. Entsprechend finden sich im eigenen Krankengut jeweils über 40 Virushepatitiden bzw. intrahepatische Schwangerschaftscholestasen. Der Bezeichnung intrahepatische Schwangerschaftscholestase ist gegenüber dem Terminus (rezidivierender) idiopathischer Schwangerschaftsikterus der Vorzug zu geben, da er das Wesentliche dieser Erkrankung charakterisiert. Der Ikterus ist zudem kein obligates Symptom. Ätiologie und Pathogenese der ICP sind im Einzelnen nicht bekannt, von einem multifaktoriellen Geschehen ist auszugehen. Diskutiert werden hormonelle, genetische und noch zu definierende Umweltfaktoren (s. Kap. 1.2.4.). Für die zentrale Bedeutung der Steroidhormone lassen sich mehrere Fakten anführen: 1. Die Erkrankung beginnt bevorzugt im letzten Trimenon, der Periode
mit den höchsten Östrogen- und Progesteron-Konzentrationen. Tierexperimentelle Studien weisen darauf hin, dass Progesteron-Metaboliten sowie Östrogene und deren Konjugate den Gallensäuretransport auf hepatozellulärer Ebene durch Down-Regulation der entsprechenden Transporter zu hemmen vermögen. Das betrifft sowohl die Aufnahme von Gallensäuren an der basolateralen Membran aus der Pfortader als auch den Export von unkonjugierten und konjugierten Gallensäuren durch die ATP-abhängigen kanalikulären Exportpumpen. Ebenso dürfte bei entsprechend prädisponierten Schwangeren den zum Ende der Schwangerschaft etwa tausendfach erhöhten Steroiden (Progesteron, Östrogene, Kortikosteroide) eine mitauslösende Rolle zukommen. So findet sich bei der Schwangerschaftscholestase ein erhöhter Anteil von sulfatierten Progesteronmetaboliten im Serum, möglicherweise Folge einer gesteigerten Synthese und eines selektiven gestörten kanalikulären Transports (6, 62, 63, 104, 117, 124, 149, 152, 153, 183, 189). Die überzufällig häufige positive Familienanamnese und das vermehrte Vorkommen in bestimmten Populationen und Regionen (Südamerika, Nordeuropa) sprechen auch für eine genetische Prädisposition bei einem Teil der Schwangeren mit ICP. In Mitteleuropa, Australien und Nordamerika liegt die Prävalenz bei 0,2 – 0,5 %, in Schweden und Finnland ist sie mit 1 – 2 % etwas höher. Die höchsten Prävalenzraten wurden in Chile und Bolivien mit 9 – 14 %, in bestimmten indianischen Populationen sogar bis 27 % beobachtet. Seit etwa 30 Jahren findet sich jedoch in Chile eine fallende Tendenz auf 4 – 6, 5 %, wahrscheinlich als Folge von Umwelt- und Ernährungseinflüssen (104, 124, 153). Als mögliche Ursachen dieser genetisch bedingten Cholestase wurden verschiedene Mutationen der kanalikulären Transportpumpen für Gallensäuren (BSEP, ABCB11-Gen) und Phosphatidylcholin (MDR3, ABCB4-Gen) sowie des FIC1-(ATP8B1-)Gens nachgewiesen (Tab. 1.7). Bei ICP-Patientinnen fanden sich bisher über 10 Mutationen im ABCB4-Gen. Unterschiedlich ausgeprägte Verminderungen der Phospholipidkonzentrationen in der Galle führen bei einigen Schwangeren möglicherweise zur Schädigung der Gallengänge durch die toxischen Effekte hydrophober Gallensäuren. Entsprechend können die Aktivitäten der Gamma-GT im Serum wie auch bei dem pädiatrischen Krankheitsbild der progressiven familiären intrahepatischen Cholestase Typ 3 (PFIC-3), die ebenfalls durch Mutationen im ABCB4-Gen verursacht wird, erhöht sein. Allerdings stellt dies nur eine Variante dieses Cholestase-Syndroms dar, da die meisten ICP-Patientinnen mit Mutationen im ABCB4-Gen, aber auch im ATP8B1-Gen normale Gamma-GT-Werte aufweisen (17, 44, 83, 90, 108, 125, 130, 135, 138, 153, 168, 171, 187). Die gleichen ABCB4-Gendefekte wie bei ICP und PFIC-3 sind auch über ein verändertes Cholesterin-Phospholipid-Verhältnis mit der Bildung von Cholesteringallensteinen assoziiert. Diese Mutationen sind somit als weiterer pathogenetischer Faktor für die erhöhte Prävalenz (bis zu 22 %) einer Cholelithiasis bei ICP-Patientinnen anzusehen (83, 104, 124, 168). Des Weiteren wurden auch bei der primär biliären Zirrhose (PBC) Mutationen im ABCB4-Gen beschrieben. Diese Mutationen gehen offensichtlich bei all diesen Erkrankungen mit einer reduzierten Funktion des MDR 3-Transporters einher. Es liegt die Beschreibung einer Patientin mit ABCB4-Mutation vor, die nacheinander an Gallensteinen, ICP und PBC als MDR 3-assoziierten Krankheitsbildern erkrankte (108). Reyes et al. (150) weisen auf intestinale Permeabilitätsstörungen als einen weiteren Faktor in der Pathogenese der ICP hin. 5 von 20 ICP-Patientinnen wiesen eine Barrierestörung während und nach der Schwangerschaft auf. Entsprechend sind Auswirkungen auf den enterohepatischen Kreislauf von Hormonmetaboliten und Gallensalzen oder auf die Aufnahme von bakteriellen Endotoxinen zu diskutieren. Dieser Befund einer gesteigerten intestinalen Permeabilität bei ICP bedarf weiterer Klärung an größeren Kollektiven. Das gleiche trifft für die Frage zu, ob eine Hepatitis C-Infektion das Risiko für die Entwicklung einer ICP über die Down-Regulation kanalikulärer Exportpumpen erhöht. Eine Hepatitis C wird häufiger bei ICP-Patientinnen als bei gesunden Schwangeren nachgewiesen (106, 137, 158). Offensichtlich ist zur Manifestation der ICP ein Überlappen von vererbten Ursachen (spezifische Mutationen hepatobiliärer Transportproteingene, erhöhte Darmpermeabilität) und erworbener Kofaktoren (hormonelle Umstellung in der Schwangerschaft, Medikamente, Infekte, Selenmangel in der Nahrung, saisonale Einflüsse mit einer Häufung im Winter etc.) notwendig. Die ICP tritt bei entsprechend prädisponierten Graviden ohne Bevorzugung irgendeiner Altersgruppe auf. Das erste und klinisch führende Symptom der ICP, das bei 64 % im 3., bei 26 % im 2. und nur selten im 1. Trimenon beginnt, ist ein meist intensiver Pruritus, der den Stamm oder die Extremitäten oder beides zugleich befallen kann. Der Juckreiz nimmt in der Regel nachts an Intensität zu und kann bei längerer Dauer zu einer erheblichen Beeinträchtigung des Allgemeinbefindens der Schwangeren führen. Über weitere Allgemeinsymptome, insbesondere gastrointestinale Beschwerden, klagen etwa 20 – 30 % der Patientinnen (69, 70, 74, 76, 77, 104, 124, 149, 153). Die Ursache des Pruritus bei ICP wie auch bei anderen cholestatischen Leberkrankheiten ist weiterhin nicht geklärt. Nach Stimulation markloser, sensorischer C-Nervenfasern in der Haut wird der Juckreiz nach Reizfortleitung über Rückenmark und Thalamus im Gyrus postcentralis der sensomotorischen Hirnrinde wahrgenommen. Die Gallensäurespiegel im Serum wie in der Haut korrelieren nicht mit der Intensität des Juckreizes und in Spätstadien cholestatischer Lebererkrankungen kann sogar trotz weiterer Erhöhung der Serumgallensäuren der Pruritus abnehmen. Somit bleibt eine pathogenetische Bedeutung der Gallensäuren offen, auch eine auslösende Rolle von Histamin ist nicht belegt. Dagegen scheint den endogenen Opioiden und wahrscheinlich auch dem Serotonin eine Mediatorfunktion zuzukommen(156,197). Wie die intrahepatische Schwangerschafts-cholestase gehen verschiedene cholestatische Syndrome, die auch in der Schwangerschaft zur Beobachtung kommen können und entsprechend abgegrenzt werden müssen, mit Pruritus einher (Tab. 1.9). Tab. 1.9 Pruritus sine materia bei cholestatischen Lebererkrankungen
Tab. 1.10 Pruritus bei schwangerschaftsspezifischen
Dermatosen
Der vorwiegend generalisierte Pruritus tritt hier primär auf unveränderter, nicht entzündlicher Haut auf (ausgenommen durch Kratzen induzierte Läsionen). Dagegen sind lokale Entzündungsmechanismen als Auslöser des Pruritus bei den spezifischen Schwangerschaftsdermatosen, die sich überwiegend im 2. und 3. Trimenon manifestieren, verantwortlich (Tab. 1.10). Im Vergleich zu den häufiger meist im 3. Trimenon auftretenden rumpfbetonten pruritischen urtikariellen Papeln und Plaques in der Schwangerschaft (PUPPP) sind die übrigen Schwangerschaftsdermatosen selten. Letztlich gilt auch für die Schwangerschaft, dass beim Auftreten eines lokalisierten oder generalisierten Pruritus eine Vielzahl von Systemerkrankungen oder Dermatosen ursächlich in Betracht zu ziehen ist (156, 197). Dem Pruritus folgt bei der ICP gewöhnlich nach 1 – 2 Wochen ein Ikterus, der bei etwa 20 % der Patientinnen deutlicher sichtbar wird. Etwa bei einem Drittel der Graviden bleibt die Gelbsucht aus, so dass man eine anikterische Verlaufsform, den Pruritus gravidarum, von einer ikterischen Verlaufsform unterscheiden kann. Nach der Geburt verschwinden zunächst der Juckreiz und dann die Gelbsucht innerhalb kurzer Zeit, spätestens nach 4 Wochen (69, 70, 74, 76, 77, 104, 124, 149, 153). Das Leberpunktat zeigt histologisch eine unregelmäßig verteilte fokale Cholestase mit Gallethromben in erweiterten Gallenkapillaren und gelegentlich auch mit Gallepigment in benachbarten Leberzellen. Elektronenmikroskopisch lässt sich darüber hinaus zeigen, dass in den dilatierten Gallenkapillaren die Mikrovilli rarefiziert oder nicht mehr nachweisbar sind. Und auch die bereits für das letzte Schwangerschaftsdrittel einer normalen Schwangerschaft recht typischen mitochondrialen Alterationen wie Größenzunahme, Verformung und kristalline Einschlüsse können noch häufiger und ausgeprägter vorhanden sein (Abb. 1.10 und 1.11). Nach der Schwangerschaft bilden sich diese Veränderungen innerhalb von 12 Wochen zurück (69, 70). Kristalline Einschlüsse finden sich in etwa 10 % in der mitochondrialen Matrix derartiger Megamitochondrien. Es handelt sich bei diesen kristalloiden Innenstrukturen um zahlreiche parallel verlaufende Membranen mit einem Durchmesser von etwa 10 nm und einem jeweiligen Abstand von etwa 20 nm, wobei Zusammensetzung und Funktion nach wie vor nicht geklärt sind (69, 70, 74, 100). Der Nachweis gleichartiger Veränderungen auch nach Langzeiteinnahme von oralen Kontrazeptiva (in höherer Dosierung), bei Chorionepitheliom und Blasenmole lässt einen Steroideinfluss vermuten. Allerdings ist dieses morphologische Bild nicht spezifisch, sondern findet sich auch außerhalb der Schwangerschaft bei verschiedenen Lebererkrankungen wie alkoholischer und nichtalkoholischer Steatohepatitis, M. Wilson sowie kryptogener Zirrhose. Offensichtlich stellen die Riesenmitochondrien in ihrer großen Formenvielfalt und den kristallinen Einschlüssen Anpassungsreaktionen der Leberzelle auf unterschiedliche Noxen dar, die insbesondere die Fettsäurenoxidation und -synthese tangieren. Die ß-Oxidation in der Mitochondrienmatrix, dem wichtigsten Weg des Fettsäurenabbaus, weist dabei eine enge räumliche und funktionale Beziehung zum Zitratzyklus und zur Atmungskette auf (s. Kap. 1.3.3.). In Übereinstimmung mit diesen morphologischen Befunden weisen die klinisch-chemischen Parameter die Konstellation einer Cholestase auf. Von den Retentionsparametern Bilirubin und Gallensäuren ist die Bestimmung der Gallensäuren im Serum der sensitivste Parameter. Ein Anstieg der Gallensäuren im Serum über 10 µmol/l kann in Einzelfällen der einzige pathologische Laborbefund sein. Daher sollte bei einem Pruritus in der Schwangerschaft und einem normalen Routinelabor (z.B. normale γ-GT-Werte) eine Messung der Gallensäuren im Serum erfolgen. Ist das fakultative Symptom des Ikterus vorhanden, liegt der Bilirubin-Spiegel zumeist unter 6 mg/dl. Die Aktivität der γ-GT, die bereits in der normalen Schwangerschaft tiefnormale Werte aufweist, bleibt überwiegend normal und zeigt nur in 10 – 15 % einen Anstieg. In diesen Fällen können Mutationen im ABCB 4 (MDR3)-Gen vorliegen (Tab. 1.7). Aufgrund der Bildung plazentarer
Abb. 1.10 Intrahepatische Schwangerschaftscholestase: Im Zentrum geringfügig erweiterte Gallenkanälchen mit Rarefizierung der Mikrovilli und Gallepigment im Lumen. Vergr. 23000fach
Abb. 1.11 Intrahepatische Schwangerschaftscholestase: Riesenmitochondrion mit heller, aufgelockerter Matrix und kristallinen Einschlüssen. Geringgradige Vakuolisierung des endoplasmatischen Retikulums. Vergr. 23000fach. Isoenzyme sind Aktivitätserhöhungen von AP und LAP schwierig zu interpretieren. Die Aktivitäten von GOT und GPT sind normal oder leicht bis mäßig erhöht (bis zu 250 U/l), nur in Einzelfällen finden sich höhere Werte. Hier muß differentialdiagnostisch das Vorliegen einer akuten Virushepatitis oder einer toxischen Leberschädigung erwogen werden. Keine Veränderungen zeigen die Aktivitäten der Gesamt-LDH (ausgenommen ein fakultativer Anstieg perinatal) und der GLDH, während die CHE gelegentlich noch deutlicher absinken kann als bereits in der normalen Gravidität. Innerhalb von 3 Wochen nach der Geburt kehren die Aktivitäten von GOT, GPT, CHE, Gesamt-LDH allmählich zur Norm zurück, bei der AP kann dieser Zeitraum bis zu 12 Wochen betragen (Abb. 1.12) (44, 69, 70, 74, 76, 104, 124, 153, 202).
Abb. 1.12 Verlauf des Bilirubins und der Enzymaktivitäten im Serum bei intrahepatischer Schwangerschaftscholestase (28-jährige 1-grav., 39. SSW). Ab 31. SSW Juckreiz, ab 33. SSW Stuhlentfärbung. Normalisierung der Werte 3 Wochen postpartal.
Abb. 1.13 Quantitative Veränderungen der Serumeiweißfraktionen in der Elektrophorese in der 40. Woche und 5 Tage post partum bei intrahepatischer Schwangerschaftscholestase (32-jährige II para). Die Prothrombinzeit (Quick-Wert) ist normal oder nur leicht verlängert. Nur bei länger dauernder Cholestase fällt die Prothrombinzeit deutlich pathologisch aus, da durch die mangelhafte Vitamin K-Resorption die Koagulationsfaktoren nur ungenügend synthetisiert werden. Auch der bereits in der normalen Schwangerschaft nachzuweisende Anstieg der Lipidfraktionen im Serum ist bei der intrahepatischen Schwangerschaftscholestase noch stärker ausgeprägt. Gesamtcholesterin, Phospholipide, Triglyzeride, VLDL und LDL sind vermehrt, HDL dagegen vermindert. Ebenfalls wird Lipoprotein X nachweisbar. In der Lipoproteinelektrophorese findet sich eine Typ IIb- oder Typ IV-Hyperlipoproteinämie (nach Fredrickson). Die intrahepatische Schwangerschaftscholestase ist dadurch charakterisiert, dass die Cholestase klinisch durch den Pruritus sehr deutlich wird, klinisch-chemisch dagegen mäßig und morphologisch nur sehr gering in Erscheinung tritt. Diese Befunde sind zwar typisch, ein eigentlich beweisendes Symptom existiert jedoch nicht, so dass die Diagnose in Einzelfällen schwierig sein kann. Es müssen verschiedene andere Erkrankungen vorwiegend mit Cholestase-Syndrom, die zum Teil eine recht unterschiedliche Ätiologie aufweisen, ausgeschlossen werden (Tab. 1.9). Neben den in Tab. 1.9 aufgeführten Erkrankungen sind weiterhin cholestatische Verlaufsformen einer akuten Virushepatitis und die ohne Pruritus einhergehenden funktionellen Hyperbilirubinämien zu nennen. Auf eine diagnostische Leberbiopsie kann in der Regel verzichtet werden. Die mütterliche Prognose ist günstig, die ICP hinterlässt trotz hoher Rezidivneigung von 45 – 70 % keine bleibenden Leberschäden. Es besteht allein eine Gefährdung durch einen vermehrten uterinen Blutverlust bei Abfall der Vitamin K-abhängigen Gerinnungsfaktoren. Aufgrund des drei- bis vierfach erhöhten Risikos der ICP-Patientinnen für Cholesteringallensteine muss allerdings im Langzeitverlauf mit einer erhöhten Inzidenz von akuter Cholezystitis, akuter Cholangitis und akuter biliärer Pankreatitis gerechnet werden (46, 94, 124, 153, 202). Eingeschränkt ist dagegen die kindliche Prognose durch Wachstumsretardierung, erhöhte Frühgeburtlichkeit und erhöhte perinatale Mortalität, so dass Schwangerschaften bei ICP als Risikoschwangerschaften einzuschätzen sind (30, 46, 47, 94, 124, 136, 153, 202). Die Angaben zur Frühgeburtlichkeit variieren zwischen 12 – 60 %, im Durchschnitt dürfte die Zahl um 20 % liegen. Das verminderte Geburtsgewicht bei der ICP ist zum Teil Folge einer verkürzten Schwangerschaftsdauer, die generell bei cholestatischen Ikterusformen zu beobachten ist. Der Geburtstermin liegt im Mittel zwischen der 36. und 38. Woche. Thorling wies bereits 1955 in einer der ersten umfassenden Darstellungen der ICP auf das verminderte Geburtsgewicht hin (Abb. 1.14) (185). Das Risiko der perinatalen Mortalität (bis zu 25 %) lässt sich nur teilweise durch die Unreife infolge der verkürzten Tragezeit (10 –20 %) erklären. In 0,4 – 3,5 % kann es – sogar bei reifen Kindern – zum plötzlichen intrauterinen Fruchttod oder zum Tod unter der Geburt kommen. Als Hinweis auf eine fetale Asphyxie ist das gehäufte Auftreten von Bradykardien (bis 14 %) und mekoniumhaltigem Fruchtwasser (20 – 44 %) zu sehen. Die Gründe für die vermehrte Frühgeburtenrate wie für den plötzlichen Fruchttod sind im Einzelnen noch zu
Abb. 1.14 Geburtsgewicht in g von 34 Kindern bei ICP (graue Säulen) im Vergleich mit 39 Kindern bei normaler Schwangerschaft (weiße Säulen) (185). klären. Bisher ist wenig über die pathogenetische Rolle der erhöhten sulfatierten Progesteronmetaboliten bekannt. Es ist zu fragen, ob sie eine Hemmung der fetalen Steroidsynthese nach Passage der Plazenta bewirken. Im Vordergrund der Diskussion stehen derzeit die erhöhten mütterlichen Gallensäuren-Konzentrationen, die zu einer Akkumulation von toxischen hydrophoben Gallensäuren auf Seiten des Feten infolge einer gestörten materno-fetalen Gallensäurenbalance führen. Zum einen besteht ein verstärkter Übertritt mütterlicher Gallensäuren, zum anderen ist zusätzlich der physiologische Transfer von Gallensäuren, die vom Feten nicht metabolisiert werden können, über die Plazenta zur Mutter bei der ICP gestört. Letztlich werden, auch auf der Basis tierexperimenteller Untersuchungen, den erhöhten Gallensäurenkonzentrationen verschiedene Schädigungsmechanismen zugeschrieben: - Schädigung der Hormonproduktion der Plazenta Die ICP als Risikoschwangerschaft bedarf der intensiven ambulanten, ggf. etwa ab der 37. Woche stationären Überwachung. Nichtinvasives und invasives Monitoring (Kardiotokographie, Sonograpie, Dopplersonographie, fetale Echokardiographie, Amniozentese, Amnioskopie, fetale Mikroblutanalysen etc.) erlauben eine fortlaufende Zustandsbeurteilung des Feten, so dass akute Gefährdungen rechtzeitig erkannt und somit ein Eingreifen, wie z. B. die vorzeitige Geburtseinleitung, zum optimalen Zeitpunkt gewährleistet werden. Da deutlich erhöhte Gallensäurenkonzentrationen im mütterlichen Serum mit einer gesteigerten fetalen Komplikationsrate korrelieren (46, 62), dürfte zukünftig der Bestimmung dieses Parameters nicht nur eine diagnostische, sondern im pränatalen Monitoring auch eine prognostische Bedeutung zukommen. So wiesen in einer schwedischen Studie von 693 Frauen mit ICP, charakterisiert durch Pruritus und Nüchtern-Serumgallensäuren > 10 µmol/l, 19 % eine schwere Verlaufsform mit erhöhtem fetalen Risiko bei Gallensäurenspiegeln > 40 µmol/l und 81 % eine milde Verlaufsform ohne gesteigerte Gefährdung des Feten bei Gallensäurenspiegeln zwischen 10 – 39 µmol/l auf (47). Um geburtshilfliche Komplikationen zu vermeiden, sollte bei der intrahepatischen Schwangerschaftscholestase wöchentlich die Bestimmung der Prothrombinzeit nach Quick, noch besser die der Einzelfaktoren durchgeführt werden. Bei länger dauernder Cholestase und bei einem deutlichen Absinken des Quick-Wertes unter 60 % (bei normalem Antithrombin III) empfiehlt sich zur Blutungsprophylaxe, auch des Kindes, die Substitution von Vitamin K. Bei der ICP wurden verschiedene Substanzen mit unterschiedlichen Wirkmechanismen zur Verminderung von Pruritus und/oder Cholestase therapeutisch eingesetzt:
Colestyramin (anfangs 12 – 16 g/Tag, später 4 – 8 g/Tag) sowie alternativ Colestipol sind nichtresorbierbare Resine und wirken als Ionenaustauscherharze. Sie binden Gallensäuren, hohe Dosen führen zur Steatorrhoe infolge verminderter Fettresorption. Weiterhin ist die Resorption von fettlöslichen Vitaminen eingeschränkt, die eventuell substituiert werden müssen. Ebenso sind Arzneimittelinteraktionen bekannt, wie z.B. die verminderte Resorption von Kortikosteroiden, Antikoagulanzien, UDC, Phenobarbital etc. Übelkeit, Erbrechen, abdominelle Beschwerden und Obstipation zählen zu den unerwünschten Wirkungen dieser Substanzen, die bei etwa drei Vierteln der Graviden den Pruritus deutlich zu lindern vermögen. Dieser Effekt der Juckreizmilderung durch Colestyramin deutet auf eine enterohepatische Zirkulation des bisher nicht eindeutig identifizierten Pruritogens hin. Der Wirkmechanismus von UDC bei ICP ist komplex und noch nicht in allen Einzelheiten bekannt. UDC, eine hydrophile, nichttoxische Gallensäure, bewirkt eine Steigerung des Galleflusses, vermindert die Konzentration der hydrophoben Gallensäuren im mütterlichen Serum und verbessert den Gallensäuretransport über die Plazenta mit der Folge einer verbesserten materno-fetalen Gallensäurebalance. Zusätzlich wird eine Zytoprotektion, eine Immunmodulation und eine Verminderung der Apoptose vermittelt. In einer oralen Dosis von 10 – 15 mg/kg Körpergewicht/Tag (Tagesdosis etwa 1 g) haben sich nicht nur eine deutliche Besserung des cholestatischen Pruritus, der Leberenzymaktivitäten und des Bilirubins, sondern auch weniger Schwangerschaftsprobleme und eine bessere Prognose für die Kinder objektivieren lassen. Eine chilenische Arbeitsgruppe gehörte zu den ersten, die vor 15 Jahren diese Effekte zeigen konnte (Abb. 1.15) (30, 47, 94, 114, 136, 202).
Abb. 1.15 Effekte von UDC (15 mg/kg/Tag) bei intrahepatischer Schwangerschaftscholestase (n=8), (mod. nach 136) Hand in Hand mit diesem positiven Effekt der UDC geht eine verbesserte Pränatalmedizin, die ebenfalls zur Senkung der perinatalen Mortalität beiträgt. Da UDC nicht oder nur in geringen Mengen in der Muttermilch zu finden ist, scheint das Stillen unter der Einnahme von UDC unbedenklich zu sein. Ob eine erhöhte Dosis von 20 – 25 mg/kg Körpergewicht/Tag einen zusätzlichen Nutzen hat, müssen weitere Studien klären. Offensichtlich stellt die ICP eine milde Verlaufsform hereditärer Cholestasen dar, so dass sich in der Regel die Gabe von UDC auf die Restfunktion des Transporters günstig auswirkt. Zur Verringerung des fetalen Risikos sollte auch bei anderen Cholestase-Syndromen in der Schwangerschaft der Einsatz von UDC diskutiert werden (Tab. 1.11). Tab. 1.11 Lebererkrankungen während der Schwangerschaft, bei denen die Gabe von Ursodeoxycholsäure zu diskutieren ist
1.3.2. Präeklampsie/HELLP-Syndrom Das Auftreten von HELLP-Syndrom oder Eklampsie (tonisch-klonische Krampfanfälle) signalisiert eine besonders schwere Verlaufsform der Präeklampsie. Das Akronym HELLP steht für „hemolysis“, „elevated liver enzymes“ (mehr als das Dreifache der Standardabweichung vom Normwert) und „low platelets“ (Thrombozytopenie < 100.000/µl). Der Begriff des HELLP-Syndroms, 1982 von Weinstein (196) geprägt, ist allerdings nicht neu. Bereits 1921 wies Stahnke (180) auf Hämolyse und Thrombozytopenie im Rahmen schwerer Eklampsien hin. In der Vergangenheit wurden weitere Begriffe wie EPH-Gestose Typ B, hepatic toxemia, hepatische Eklampsie oder hepatogene Verlaufsform der EPH-Gestose zur Kennzeichnung dieser Komplikation benutzt (70, 71, 74). Schwangere mit Präeklampsie bzw. Eklampsie entwickeln in 4 – 35 % zusätzlich ein HELLP-Syndrom. Die Inzidenz des HELLP-Syndroms liegt heute bei 0,17 – 0,8 % aller Lebendgeburten, aufgrund einer verbesserten Schwangerenvorsorge sank dagegen die Eklampsierate auf jetzt 0,03 – 0,1 % aller Geburten (147, 148). Das Risiko für die Entwicklung einer Präeklampsie ist bei familiärer Belastung (Auftreten bei Mutter und/oder Schwester), bei Erstgebärenden (52 – 81 %), besonders bei jungen Erstgebärenden und Spätgebärenden (über 35 Jahre), bei Präeklampsie in früherer Schwangerschaft und bei Mehrlingsschwangerschaften erhöht. Verschiedene internistische Grunderkrankungen wie Nierenerkrankung, chronische Hypertonie, Adipositas, Diabetes mellitus, systemischer Lupus erythematodes, Sklerodermie sowie das Vorliegen thrombophiler Risikofaktoren bedingen ebenfalls ein erhöhtes Erkrankungsrisiko. Insbesondere bei Auftreten von Präeklampsie bzw. HELLP-Syndrom vor der 34. Schwangerschaftswoche sollte nach angeborenen (Protein-S- und Protein-C-Mangel, Faktor V-Leiden-Mutation u.a.) und erworbenen (Lupusantikoagulans, Anti-Phospholipid-Antikörper) Thrombophilien als prädisponierende Faktoren gefahndet werden. Die Schwangerschaftsthrombozytopenie (Grenzwert < 70.000/µl), die bei etwa 5 % der Schwangerschaften auftritt, weist ebenfalls ein erhöhtes Risiko für das spätere Auftreten eines HELLP-Syndroms auf (148). Die Ätiologie der Präeklampsie ist nicht sicher bekannt. Mehrere pathogenetische Mechanismen werden diskutiert wie eine genetische Prädisposition, angeborene und erworbene Thrombophilien, utero-plazentare Perfusionsstörungen, eine Überempfindlichkeit der Gefäße gegenüber vasopressorischen Substanzen, ein Ungleichgewicht zwischen Prostacyclin und Thromboxan A2 sowie eine pathologische Immunreaktion der Mutter gegenüber dem Feten. Als Folge dieser Störungen kommt es zu segmentalen Vasospasmen mit Erhöhung des peripheren Gefäßwiderstandes, zu Endothelläsionen sowie zu gesteigerter intravaskulärer Gerinnungsaktivierung und herabgesetzter fibrinolytischer Aktivität. Die resultierende Mikroangiopathie hat eine Schädigung der Erythrozyten mit Hämolyse zur Folge sowie einen vermehrten Thrombozytenverbrauch bzw. –umsatz (147, 148, 198). Bei leichter Präeklampsie können Mikrozirkulationsstörungen und eine subklinische chronische Verbrauchskoagulopathie vom mütterlichen Organismus voll kompensiert werden. Persistieren jedoch bei schwerer Eklampsie sowie bei HELLP-Syndrom und Eklampsie endotheliale Dysfunktion und intravasale Gerinnungsaktivierung mit Thrombozytenverbrauch kann sich innerhalb von Stunden eine disseminierte intravaskuläre Gerinnung (DIG) bis hin zur Verbrauchskoagulopathie entwickeln. Die Folgen sind Thrombosen und Mikrozirkulationsstörungen in zahlreichen Organsystemen (Gehirn, Nieren, Leber u.a.) in wechselnder Ausprägung. Es resultiert ein variables klinisches Bild einschließlich Multiorganversagen. Die Leber nimmt in der Regel bei der Präeklampsie wie auch bei der Hyperemesis gravidarum keine führende Stellung im Krankheitsverlauf ein, und es besteht auch keine sichere Korrelation zwischen dem Ausmaß der Leberbeteiligung und der Schwere der Präeklampsie. Die Leber ist somit weder obligat noch adäquat bei der Präeklampsie beteiligt. Die Gründe für diese unterschiedliche Leberbeteiligung, für eine „Prädisposition“ bestimmter Schwangerer, speziell beim HELLP-Syndrom, sind unbekannt (70, 71, 74). Klinik. Das HELLP-Syndrom tritt in der fortgeschrittenen Schwangerschaft am häufigsten zwischen der 32. bis 34. Schwangerschaftswoche, seltener in den vorhergehenden Wochen (bis 10 %) und in bis zu 30 % um die Geburt und im frühen Wochenbett auf. Bei der Art der Leberschädigung beim HELLP-Syndrom – und die Leber steht im Mittelpunkt dieses Syndroms – handelt es sich um eine akute Durchblutungsstörung in sehr unterschiedlicher Ausprägung (70, 71, 74). Bereits vor fast 50 Jahren konnte die Minderdurchblutung der Leber gezeigt werden. Während die Durchblutung bei Nichtschwangeren und gesunden Schwangeren in gleicher Weise 1830 ml/min pro 1,73 m² betrug, sank sie bei Präeklampsie auf 1017 ml/min pro 1,73 m² ab (61). Als Folge des obstruktiven intravasalen Gerinnungsprozesses kommt es zu einer Behinderung des Blutflusses und damit zur akuten Leberstauung. Entsprechend ist die Leber vergrößert und die Kapsel straff, was wiederum das klinische Leitsymptom mit Schmerzen im rechten Oberbauch (86 – 92 % der Fälle), oft begleitet von Übelkeit und Erbrechen (45 – 86 %) erklärt. In 20 – 40 % der Fälle können Oberbauchschmerzen der klinisch-chemischen Manifestation um Tage vorausgehen. Bis zu 20 % der Schwangeren haben keinen Bluthochdruck, bis zu 15 % keine oder nur eine geringe Proteinurie, bei 15 % fehlen die klinischen Zeichen der Präeklampsie vollständig (HELLP-Syndrom sine preeclampsia). In diesen Fällen können die Leitsymptome „Oberbauchschmerzen“ oder „Übelkeit und Erbrechen“ klinisch richtungweisend sein (70, 71, 147). Das HELLP-Syndrom ist am häufigsten der Auslöser von Oberbauchschmerzen, jedoch müssen verschiedene andere Erkrankungen in die differentialdiagnostischen Erwägungen einbezogen werden (Tab. 1.12). Zur Differentialdiagnose von Übelkeit und Erbrechen s. Tab. 4.3. Treffend wurde die schwere Präeklampsie als „another great imitator“ bezeichnet. Tab. 1.12 Differentialdiagnose des Oberbauchschmerzes in der Spätschwangerschaft
Diagnostik. Unabhängig von der Ausprägung der Präeklampsie-Symptomatik muss bei Schwangeren mit unklaren abdominellen Beschwerden auch an die Entwicklung eines HELLP-Syndroms gedacht werden. Im Rahmen einer intensiven geburtsmedizinischen Überwachung sollte im Verdachtsfalle das in Tab. 1.13 aufgeführte Laborscreening konsequent durchgeführt werden. Bei unklaren Befunden müssen diese klinisch-chemischen Parameter in 6- bis 8-stündlichen Abständen erneut kontrolliert werden. Ein beginnendes Syndrom kann in wenigen Stunden in das Vollbild mit der charakteristischen laborchemischen Trias übergehen. Tab. 1.13 Laborscreening bei Verdacht auf HELLP-Syndrom
Abb. 1.16 HELLP-Syndrom (22-jährige Patientin, 32. SSW). Anstieg der LDH-Aktivitäten und der Konzentration des Gesamt- Bilirubins im Serum. Je nach Ausprägung der Hämolyse findet sich ein Anstieg des überwiegend indirekten Bilirubins bis 2,5 mg/dl, aber auch höher, sowie eine Erhöhung der Aktivitäten von LDH und HBDH im Serum (Abb. 1.16). Die Haptoglobinkonzentration ist vermindert, die Retikulozytenzahl erhöht und Fragmentozyten werden im peripheren Blutausstrich nachweisbar. Postpartal normalisiert sich die Haptoglobinkonzentration, der sensitivste Hämolyseparameter, in der Regel in 1 – 2 Tagen. Bei progredienter Erkrankung kann es innerhalb weniger Stunden zu einer Thrombozytenzahlverminderung auf < 50.000/µl kommen. Mütterliches und kindliches Morbiditätsrisiko korrelieren wahrscheinlich mit dem Ausmaß der Thrombozytopenie. Neben der dynamisch abfallenden Thrombozytenzahl auf < 100.000/µl weisen ein Anstieg der D-Dimere und ein Abfall von Antithrombin III auf einen schweren Krankheitsverlauf hin. Die globalen Gerinnungsparameter wie Quick-Wert, Fibrinogen und Thrombinzeit sind dagegen weniger sensitiv. Blutungskomplikationen in verschiedenen Organen sind auf eine Thrombozytopenie hinweisende klinische Zeichen (Abb. 1.17) (Tab. 1.14). Postpartal kommt es in 20 – 30 % zu einer reaktiven Thrombozytose mit erhöhter Thrombemboliegefahr, so dass sich die Gabe von niedrigdosiertem Heparin und Acetylsalicylsäure empfiehlt (Abb. 1.18).
Abb. 1.18 HELLP-Syndrom (20-jährige Patientin, 33. SSW). Reaktive Thrombozytose Tab. 1.14 Thrombozytopenien während der Schwangerschaft
Die akute Durchblutungsstörung der Leber beim HELLP-Syndrom ist primär in der Peripherie des Läppchens lokalisiert. Minderung von Sauerstoff und akuter Mangel an Substraten führen zunächst zu einer Störung der Zellabdichtung, so dass die Enzyme entsprechend ihrem Konzentrationsgefälle in die Zirkulation austreten. Je stärker die Einzelzelle geschädigt wird und je mehr Areale in der Leber betroffen sind, umso höher ist dieser Enzymanstieg. Entsprechend erweisen sich Enzymbestimmungen im Serum auch beim HELLP-Syndrom als die sensibelsten Indikatoren einer Leberzellschädigung. Im eigenen Krankengut (Tab. 1.15) findet sich bei etwa 80 % der HELLP-Patientinnen ein leichter bis mäßiger Anstieg der Aktivitäten von GPT und GOT bis 200 U/l. Bessert sich die Situation postpartal, erlangen die Zellen ihre Integrität wieder und innerhalb von 1 – 2 Wochen kommt es zum raschen Abfall der erhöhten Enzymaktivitäten im Serum, nahezu den physiologischen Halbwertzeiten entsprechend (70, 71, 74, 77).
Diese rasche Normalisierung nach der Geburt und die höheren Werte der zytosolischen GPT gegenüber der bilokulären GOT (zu 70 % in den Mitochondrien) weisen darauf hin, dass die aktuelle Zellschädigung relativ leicht ist. Allerdings ist bei stärkeren Schäden der Aktivitätsanstieg der GOT höher als derjenige der GPT. Die engen zeitlichen Beziehungen zum Entbindungstermin werden daran deutlich, dass sich bei drei Vierteln der Schwangeren das HELLP-Syndrom präpartal manifestiert, d.h. die höchsten Aktivitäten der Transaminasen werden kurz vor und nach der Geburt gemessen (Abb. 1.19). Die Aktivität der γ-GT im Serum ist meist normal, in Einzelfällen kann sie aber auch erhöht sein (Abb. 1.20). Je nach Schwere der Einzelzellschädigung und Verminderung der Durchblutung können die Syntheseleistung und damit die Aktivität der Cholinesterase vermindert sein. Bei den seltenen schweren Verlaufsformen und/oder Schockzuständen findet man im Serum einen steilen Anstieg der Aktivitäten von GOT, GPT, LDH und besonders der mitochondrialen GLDH auf Werte von mehreren Hundert bis zu einigen Tausend U/l (70, 71, 74). Dieses Enzymmuster ist jedoch nicht pathognomonisch für die schwere Verlaufsform des HELLP-Syndroms, sondern ist Ausdruck einer uniformen Reaktion der Leber auf jede schwere Durchblutungsstörung unabhängig von ihrer Ätiologie. Hier wird die enge Integration der hepatischen in die systemische Zirkulation deutlich. So findet sich außer im Schock dieses Enzymmuster infolge akuter Durchblutungsstörung bei akuter Rechtsherzinsuffizienz, bei akuter Pfortaderthrombose, bei akutem Verschluss der Arteria hepatica und auch bei einer (speziell der gedeckten) Leberruptur, die ebenfalls zu einem hypoxischen Schaden führen kann (Abb. 1.21) (70, 71, 74). Auch das morphologische Bild kann in Abhängigkeit von Ausmaß und Dauer der Zirkulationsstörungen erheblich variieren. Histologisch charakteristisch sind in leichten Fällen Fibrinthromben in den Sinusoiden vorwiegend der Läppchenperipherie. Bei stärkerer Schädigung kann es in diesen Arealen zu Blutungen kommen, und die Zellen können einzeln oder in Gruppen nekrotisch werden. Typischerweise weisen die Fibrinablagerungen und hämorrhagischen Nekrosen eine portale Verteilung auf. Bei noch schwereren Alterationen breiten sich die hämorrhagischen Nekrosen aus; sie können ganze Läppchen erfassen bis hin zur Bildung ausgedehnter Hämatome.
Abb. 1.19 HELLP-Syndrom. Verhalten der Aktivitäten
von GOT und GPT im Longitudinalschnitt Kommt es bei der Patientin zu einem Schock, so pfropfen sich auf dieses Bild zusätzlich zentroazinäre Nekrosen auf, so dass ausgedehnte flächenhafte Nekrosen und Blutungen das Parenchym durchziehen. Entzündliche Infiltrate fehlen anfangs. Erst bei längerem Krankheitsverlauf sind Kupferzellaktivierungen sowie eine Vermehrung von polymorphkernigen Granulozyten und Rundzellen zu beobachten. Nach Überstehen der Erkrankung bilden sich diese Veränderungen ohne Restschäden zu hinterlassen zurück (Abb. 1.22). Makroskopisch ist die Leber vergrößert, die Kapsel straff. Beim Auftreten von Blutungen (Häufigkeit 2 – 2,5 %) sind diese unregelmäßig verteilt, von variabler Größe und unregelmäßiger Begrenzung. Bevorzugt liegen die Blutungen unter der Kapsel des rechen Leberlappens mit der ständig drohenden Gefahr der Ruptur (1,5 – 1,8 %) (Abb. 1.23). Bei jedem Verdacht auf HELLP-Syndrom sind für die Diagnose die bildgebenden Verfahren, vor allem die Sonographie, optional. Erste Hinweise können diffuse oder landkartenartige Änderungen in der Dichte der Leberstruktur und ein abgerundeter Leberrand sein, Zeichen, die bereits vor dem Nachweis klinisch-chemischer Veränderungen vorhanden sein können. In sonographischen Verlaufsuntersuchungen sollte es gelingen, die Entwicklung von intrahepatischen oder subkapsulären Hämatomen, einer Fettleber, einer Pankreatitis, einer pathologischen Flüssigkeitsansammlung im Bauchraum, eines Pleuraergusses oder einer Cholelithiasis nachzuweisen oder auszuschließen
Abb. 1.20 HELLP-Syndrom. Verhalten der Aktivität
der γ-GT im Longitudinalschnitt (n=31) (Normwert < 18 U/l)
Abb. 1.21 Verlauf der Aktivitäten von GOT, GPT, GLDH im Serum bei Spontanruptur eines subkapsulären Hämatoms in der 27. Schwangerschaftswoche bei Präeklampsie
Abb. 1.22 HELLP-Syndrom. Läppchenperiphere Fibrinausfällungen
Abb. 1.23 HELLP-Syndrom. Subkapsuläre Hämatombildungen Der Verlauf des HELLP-Syndroms ist im Einzelfall nicht vorhersehbar. Es gibt schleichende Verläufe mit sehr langsamer Entwicklung, intermittierende Verläufe mit spontaner Remission und Rückbildungen der pathologischen Laborparameter unter medikamentöser Therapie mit der Gefahr erneuter Schübe. Dagegen stehen foudroyante Verläufe mit teils irreversiblen Schäden, die in wenigen Stunden aus einer Präeklampsiesymptomatik hervorgehen. Die Zahlen für das Wiederholungsrisiko für ein HELLP-Syndrom variieren zwischen 2,1 und 19 %, sie liegen in Deutschland bei 14 % (147). Mit Komplikationen muss beim HELLP-Syndrom häufiger als bei der Eklampsie gerechnet werden: - Entwicklung einer disseminierten intravaskulären Gerinnung/Verbrauchskoagulopathie
(bis 38 %) Die gastroenterologischen Komplikationen beinhalten Aszitesbildung (bis 8 %), gastrointestinale Blutungen, die seltene Kombination mit einer akuten Schwangerschaftsfettleber, intrahepatische Blutungen (bis 2,5 %) und als gefährlichste Komplikation die Leberruptur (bis 1,8 %). Die mütterliche Sterberate beim HELLP-Syndrom liegt weltweit bei etwa 3 %. Die kindliche Mortalität beträgt 7,7 – 37 %. Die hohe fetale Gefährdung ist einmal bedingt durch vorzeitige Plazentalösung und intrauterine Asphyxie vor allem als Folge von chronischer Plazentainsuffizienz mit intrauteriner Wachstumsretardierung und ergibt sich zum anderen aus der Frühgeburtlichkeit, die aus der sofortigen Schwangerschaftsbeendigung bei schwerem Verlauf insbesondere vor der 34. Schwangerschaftswoche resultiert (147, 198). Große differentialdiagnostische Schwierigkeiten bereiten schwere Verlaufsformen des HELLP-Syndroms, die ein Multiorganversagen mit disseminierter intravaskulärer Gerinnung und Verbrauchskoagulopathie aufweisen, gegenüber anderen akut ikterischen Krankheiten mit generalisierter Organbeteiligung am Ende der Schwangerschaft (Tab. 1.16). Trotz unterschiedlicher Ätiologie gibt es große klinische Ähnlichkeiten. Da die Schwangerschaft bei diesen Multisystemerkrankungen (ausgenommen akute Hepatitis und M. Wilson) ein auslösender Faktor ist, sollte eine rasche Beendigung der Schwangerschaft angestrebt werden. Nur in diesen Fällen, wenn Unklarheit besteht, ist die Leberpunktion (wenn möglich transjugulär) zu diskutieren. Ansonsten ist die Leberpunktion nicht Bestandteil der Diagnostik beim HELLP-Syndrom. Tab. 1.16 Differentialdiagnose akuter ikterischer Krankheiten mit generalisierter Organbeteiligung in der Schwangerschaft
Eine kausale Therapie von Präeklampsie/HELLP-Syndrom, abgesehen von der Entbindung, existiert bis heute in Ermangelung eines gesicherten pathophysiologischen Gesamtkonzepts nicht. Ebensowenig ist eine Prophylaxe mit medikamentösen und diätetischen Maßnahmen sicher möglich. Die Behandlung ist daher bis jetzt lediglich eine symptombezogene Therapie zur Stabilisierung der Erkrankung bis zur Geburt. Meist im Rahmen eines intensivmedizinischen Managements konzentriert sich die Therapie auf blutdrucksenkende und auf antikonvulsive Maßnahmen sowie auf die Korrektur von Hämostasestörungen. Weitere Untersuchungen müssen zeigen, ob die systemische Gabe von Glukokortikoiden nicht nur die Induktion einer Lungenreife, sondern auch eine Stabilisierung selbst schwerer HELLP-Syndrome ermöglicht (147). Bei schweren Verläufen ist nach der 34. Schwangerschaftswoche möglichst rasch die Entbindung als einzig verfügbare kausale Therapie anzustreben. Eine Entbindung vor der 34. SSW geht häufig mit erheblicher Frühgeburtlichkeit einher. Ob hier mit einem expektativem Vorgehen die erhöhte neonatale Morbidität und Letalität verringert werden kann, muss in einem Perinatalzentrum abhängig vom mütterlichen und fetalen Zustand individuell entschieden werden. Unmittelbare therapeutische Konsequenzen ergeben sich aus der Leberbeteiligung bei HELLP-Syndrom nicht, hier ist auf die Entstehung subkapsulärer Hämatome mit der Gefahr der Leberruptur zu achten. 1.3.3. Anhang: Leberruptur, intraabdominelle Blutung Seit der Erstbeschreibung im Jahre 1844 wurden bis heute ca. 200 Fälle von spontaner Leberruptur in der Schwangerschaft beschrieben. Diese schwere, lebensbedrohliche Komplikation ereignet sich überwiegend im letzten Trimenon, gelegentlich im Puerperium und nur ausnahmsweise in der frühen Schwangerschaft. Überwiegend betroffen waren ältere Mehrgebärende im Alter zwischen 35 und 45 Jahren, die in über 75 % der Fälle eine Gestose-Symptomatik aufwiesen. Ein zunehmender Ikterus, eine sich vergrößernde Leber, eine Leukozytose und Anämie sollten den Verdacht auf eine intrahepatische oder subkapsuläre Hämatombildung lenken. Tritt eine Ruptur ein, ist das klinische Bild geprägt von akuten abdominellen Schmerzen, besonders im rechten Oberbauch, und einem rasch sich entwickelnden Schock. Entscheidend für das Überleben der Mutter ist die sofortige Diagnosestellung (bildgebende Verfahren, Arteriographie, Blutaspiration aus der Bauchhöhle) und die sich unmittelbar anschließende therapeutische Intervention. Je nach Schwere der Leberverletzung müssen unterschiedliche operative Verfahren eingesetzt werden, wobei das Spektrum von der notfallmäßigen Tamponade (Packing) bis zur orthotopen Lebertransplantation reichen kann. Ätiologisch werden verschiedene Faktoren diskutiert, die sich wahrscheinlich kombinieren. Einmal der schwangerschaftsbedingte erhöhte intraabdominelle Druck, der eine zusätzliche Steigerung durch Erbrechen, Krämpfe und den Geburtsvorgang erfährt, und zum anderen die Gefäßläsionen und die Nekrosen in der Leber mit der sich ausbildenden hämorrhagischen Diathese. Wird im Rahmen des HELLP-Syndroms ein subkapsuläres Hämatom ohne Ruptur, bevorzugt lokalisiert im rechten Leberlappen, nachgewiesen, kann eine abwartende Haltung unter strenger klinischer Kontrolle eingenommen werden. Bei reifem Kind wird ein chirurgisches Eingreifen empfohlen mit gleichzeitiger abdominaler Schnittentbindung. Dieses differenzierte Vorgehen in Verbindung mit Fortschritten in Diagnostik und Therapie hat dazu geführt, daß die mütterliche und kindliche Mortalität auf etwa 30 % gesenkt werden konnte. Ausnahmsweise finden sich auch Einblutungen in die Leber und Leberrupturen in der Spätschwangerschaft bei Frauen mit akuter Schwangerschaftsfettleber (70, 120). Nicht in jedem Falle liegt jedoch der Leberruptur eine schwangerschaftsspezifische Lebererkrankung zugrunde. In Einzelfällen bestand allein ein arterieller Bluthochdruck oder aber es fand sich keine auslösende Ursache. Aber auch so seltene Erkrankungen wie das kavernöse Leberhämangiom, der Amöbenabszeß, das primäre Leberkarzinom und insbesondere das Leberzelladenom können in der Schwangerschaft rupturieren (s. Kap. 1.2.11.). Im Rahmen einer Bauchhöhlenschwangerschaft erfolgt die Nidation des Eies auf dem Peritoneum, vor allem des Darmes, des Omentum, der Uterusrückwand, aber auch auf der Leber. Gewöhnlich ist die Plazenta auf der Unterfläche des rechten Leberlappens lokalisiert. Meist enden extrauterine Graviditäten durch Komplikationen wie intraabdominelle Blutung oder Perforation, gelegentlich werden sie aber auch ausgetragen. Somit kann auch eine „Leberschwangerschaft“ zum Ausgangspunkt einer intraabdominellen Blutung werden. Weltweit sind erst 15 Fälle bekannt geworden, in denen eine Leberschwangerschaft erfolgreich durch Schnittentbindung beendet werden konnte. Beim plötzlichen Auftreten eines akuten Abdomens mit den Zeichen des hämorrhagischen Schocks kommen weitere, nicht von der Leber ausgehende Blutungsursachen in Frage. Aus dem Bereich des Oberbauches seien noch Blutungen aus der Milz und den Milzgefäßen nach spontaner Ruptur genannt. Ein besonderes Problem stellt die Ruptur arterieller Aneurysmen dar, die bei mehr als der Hälfte aller Rupturen bei Frauen in der Schwangerschaft oder im Wochenbett auftreten. Jeweils bis zu 100 Fallbeobachtungen rupturierter Aneurysmen liegen für Aorta, Hirngefäße, Koronarien, Ovarialgefäße und Nierenarterien vor. Als auslösende pathogenetische Mechanismen werden u.a. Mediaauflockerung, Intimahyperplasie und erhöhtes Herzzeitvolumen angenommen. Unter den Aneurysmen viszeraler Arterien, die seit dem Einsatz sonographischer Untersuchungen vermehrt als Zufallsbefunde gesehen werden, finden sich bei jungen Frauen gehäuft Aneurysmen der Arteria lienalis. In der Schwangerschaft erhöht sich die Rupturrate dieser Aneurysmen signifikant. 25 – 45 % rupturieren im 3. Trimenon oder früh postpartal mit einer mütterlichen Letalität von 75 % und einer fetalen von 95 %. Gefährdet sind vor allem Mehrgebärende (98). Die therapeutische Konsequenz ist die frühzeitige Therapie von Aneurysmen, auch im asymptomatischen Stadium. Bei stattgehabter Ruptur ist die Notfalloperation die einzige therapeutische Konsequenz. 1.3.4. Akute Schwangerschaftsfettleber (ASFL) Die ASFL ist eine seltene schwangerschaftsspezifische Komplikation überwiegend des letzten Trimenon, die sich mit Symptomen des akuten Leberversagens (ALV) präsentiert. Eine vorbestehende chronische Lebererkrankung liegt nicht vor. Die Ursachen eines ALV sind vielfältig, am häufigsten liegen virale Infektionen oder medikamentös-toxische Schäden mit massivem Leberparenchymuntergang vor. Bei der ASFL finden sich jedoch keine ausgedehnten Parenchymnekrosen, sondern zum Ausfall der Leberfunktion führt hier eine mikrovesikuläre Verfettung der Leberzellen. Entsprechend ist der Gehalt an Triglyzeriden und freien Fettsäuren in der Leber bei ASFL erhöht (36). Allen Ursachen des ALV gemeinsam ist, dass sie in einer gemeinsamen Endstrecke, dem klinischen Bild des ALV, enden mit einem breiten Spektrum klinischer, klinisch-chemischer und neurophysiologischer Veränderungen. Somit ist die ASFL als Multisystemerkrankung mit variabler Symptomatik anzusehen, wobei die Leber zunächst im Mittelpunkt dieses Syndroms steht. Entsprechend weist die Klinik zahlreiche Gemeinsamkeiten mit schweren Verlaufsformen des HELLP-Syndroms (Tab. 1.16), aber auch mit dem Bild der Sepsis auf. Wahrscheinlich stammt die erste histopathologische Beschreibung der ASFL von Tarnier 1857. Das klinische Bild wurde erstmalig 1934 von Stander und Cadden beschrieben. Die erste vollständige Darstellung der Erkrankung gab Sheehan 1940, der in ihr eine nosologische Entität erkannte und sie als „akute gelbe Leberatrophie der Schwangerschaft“ bedingt durch eine mikrovesikuläre Verfettung der Hepatozyten histologisch von nekrotisierenden Verlaufsformen verschiedener Lebererkrankungen abgrenzte. Ober und LeCompte prägten 1953 den Begriff „akute fettige Metamorphose der Leber in der Schwangerschaft“ und grenzten die Erkrankung damit auch nomenklatorisch von nekrotisierenden Lebererkrankungen in der Schwangerschaft ab. Schultz et al. und Kunelis et al. beschrieben 1963 und 1965 ein weitgehend gleiches Syndrom nach hochdosierten Tetrazyklingaben in der Schwangerschaft. Dies führte zur Abgrenzung einer „Tetrazyklin-assoziierten Schwangerschaftsfettleber“ von einer „idiopathischen Schwangerschaftsfettleber“ (69, 70, 74, 151). Verlässliche Angaben zur Inzidenz der weltweit in allen ethnischen Gruppen auftretenden ASFL fehlen, Zahlen zwischen 1:9000 und 1:13.000 dürften am ehesten zutreffen. Die bisher sicherste Berechnung stammt aus dem Jahre 1984 mit einer relativen Häufigkeit von einer Erkrankung auf 13.328 Schwangerschaften. Diese Angabe stützt sich auf die Beobachtung von ungefähr 120.000 Geburten in 10 Jahren in Los Angeles (141). Allerdings sind derartige Angaben kritisch zu sehen. Milde Verlaufsformen dürften am ehesten nicht erkannt, schwere Verlaufsformen vor allem mit dem HELLP-Syndrom verwechselt werden. Da ein diagnostisch beweisender Marker nicht existiert, kann die Diagnose einer ASFL sicher nur gestellt werden, wenn sie morphologisch bestätigt ist. Aus diesem Grunde beziehen wir uns in unseren Aussagen im Wesentlichen auf histologisch gesicherte Erkrankungsfälle. Das sind zum einen 325 bioptisch bestätigte Fälle von insgesamt 400 Erkrankungen, über die bis 1994 berichtet wurde und von denen bei 245 Patientinnen detaillierte klinische Angaben vorliegen (151), sowie zum anderen 6 eigene Beobachtungen, von denen 4 histologisch gesichert sind. Von diesen kamen 3 Erkrankte ad exitum, einmal infolge septischen Schocks, zweimal im akuten kardiovaskulären Versagen (69, 70, 151, 173).
Abb. 1.24 Altersverteilung bei 240 Graviden mit bioptisch gesicherter ASFL
Abb. 1.25 Verteilung von Erst- und Mehrgebärenden bei 217 Graviden mit bioptisch gesicherter ASFL Die ASFL kann in jedem Alter auftreten, die jüngste Patientin war 16 Jahre alt, die älteste 46, das Durchschnittsalter liegt bei 27 Jahren. Erstgebärende erkranken mit 42 % häufiger als Mehrgebärende (Abb. 1.24 und 1.25). Die Erkrankung entwickelt sich fast immer im 3. Trimenon zwischen der 29. und 40. Woche, im Durchschnitt in der 35. Woche. Fälle mit Beginn in der 24. Schwangerschaftswoche sowie postpartal früh im Wochenbett stellen Ausnahmen dar (69, 70, 74, 151). Ätiologie und Pathogenese der ASFL sind bisher nicht vollständig geklärt. Zwar sind in den vergangenen Jahren einzelne pathophysiologische Mechanismen herausgearbeitet worden, die ein Glied bilden könnten in der pathogenetischen Kette, die schließlich in das charakteristische morphologische Bild der ASFL einmündet, der ätiopathogenetische Prozess als ganzer ist jedoch zur Zeit noch überwiegend hypothetisch. Die ASFL steht in einer Reihe mit anderen Erkrankungen, die ebenfalls bei unterschiedlichen auslösenden Faktoren letztlich eine mikrovesikuläre Verfettung als uniforme Reaktion der Leber aufweisen können. Da nur einzelne Aspekte der Pathogenese bekannt sind, sollte man die ASFL derzeit noch zur idiopathischen Untergruppe zählen (Tab. 1.17). Für die Entwicklung der hepatischen Steatose wird einer mitochondrialen Dysfunktion eine zentrale Rolle zugeschrieben. Medikamente, Alkohol, Zytokine oder Sexualhormone können die mitochondriale DNA (die mitochondrialen Struktur- und Funktionsproteine werden außerdem durch die chromosomale und vor allem durch die nukleäre DNA kodiert) wie das Coenzym A (z. B. Salizylate, Valproinsäure) schädigen, die Enzyme der mitochondrialen Fettsäureoxidation hemmen (z. B. Tetrazykline, Glukokortikoide, Valproinsäure), direkt in die Atmungskette eingreifen (z. B. TNF-a, NO, Endotoxine) oder vermehrt die in der inneren Mitochondrienmembran lokalisierten Permeabilitäts-Poren öffnen (z. B. Salizylate, Gallensäuren, TNF-a, Valproinsäure). Einzelne Pharmaka (z. B. Valproinsäure) können dabei mehrere Effekte auf die mitochondriale Funktion aufweisen (140). Da akute Störungen der Beta-Oxidation der Fettsäuren eine mikrovesikuläre Verfettung verursachen, steht in der Pathogenese der ASFL offensichtlich eine Beeinträchtigung dieser mitochondrialen Funktion im Vordergrund. Die Verstoffwechselung der Fettsäuren erfolgt in den Mitochondrien überwiegend durch Beta-Oxidation. Hierzu müssen die langkettigen Fettsäuren (C14 bis C20) mit Hilfe des Carnitin-Shuttles aktiviert und über die mitochondriale Innenmembran als Carnitinester in die Matrix transportiert werden. Die kurz- und mittelkettigen Fettsäuren (C4 bis C12) diffundieren frei durch die mitochondriale Doppelmembran. Bei der Beta-Oxidation werden die aktivierten Fettsäuren zu Acetyl-CoA abgebaut. Während Acetyl-CoA in den Zitratzyklus eingeschleust wird, werden die Reduktionsäquivalente FADH2 und NADH zur Energiegewinnung in Form von ATP in der Atmungskette oxidiert. Werden aufgenommene oder synthetisierte Fettsäuren in der Leberzelle nicht zur Energiegewinnung herangezogen, können sie nach Reveresterung als Triglyzeride gespeichert oder in Form von VLDL ausgeschleust werden. Tierexperimentell (Maus, Ratte) führte die Gabe von weiblichen Sexualhormonen wie auch die späte Schwangerschaft selbst nicht nur zur Verminderung der Fettsäureoxidation und zur Hemmung der Aktivität des Tricarbonsäurezyklus, sondern auch zu morphologischen Änderungen der mitochondrialen Ultrastruktur (55). Auch bei der ASFL können wie in der normalen Schwangerschaft und bei der ICP Größenzunahme und Verformung der Mitochondrien vorhanden sein. Hieraus kann sich eine Beeinflussung der mitochondrialen Funktion, aber auch eine Störung des Zusammenspiels der Stoffwechselwege in den kommunizierenden Funktionsräumen ergeben. Die besonders bei der ICP auftretenden mitochondrialen Alterationen prädisponieren offensichtlich aber nicht zur häufigeren Entwicklung einer ASFL, das gemeinsame Auftreten in Einzelfällen muss als zufälliges Zusammentreffen interpretiert werden (70, 74). Tab. 1.17 Lebererkrankungen mit mikrovesikulärer Fettspeicherung
Die physiologische Hypertriglyzeridämie in der Schwangerschaft mit einem Maximum im 3. Trimenon ist Folge einer vermehrten VLDL-Synthese und verminderten Lipoproteinlipase-Aktivität. Östrogene spielen hier in der Regulation eine besondere Rolle. Sie bewirken eine gesteigerte Lipolyse im Fettgewebe mit konsekutiver erhöhter Aufnahme von freien Fettsäuren in die Leber und eine gesteigerte VLDL-Synthese, aber auch eine Behinderung der mitochondrialen Beta-Oxidation. Zu klären ist, unter welchen Bedingungen (z. B. sehr hohe Konzentrationen) Östrogene an dem Auftreten akuter Störungen der Beta-Oxidation beteiligt sind, zu fragen ist aber auch nach den Effekten von Insulin und HPL, denen gleichfalls im Verlauf der Schwangerschaft eine Schlüsselfunktion in der Regulation der Triglyzeride zukommt. Entwickelt sich z. B. eine Insulinresistenz und können erhöhte Insulinspiegel ebenfalls die mitochondriale Beta-Oxidation behindern. Weiterhin ist bisher nicht bekannt, ob den extramitochondrialen Oxidationswegen (in Peroxisomen und mikrosomal) in der Situation eines erhöhten Angebots von freien Fettsäuren an die Leberzelle und/oder einer gestörten mitochondrialen Beta-Oxidation in der Pathogenese der ASFL eine Bedeutung zukommt. Zu prüfen ist weiterhin, ob die Immunreaktionen z. B. durch Mikrochimärismus bei der ASFL (Fetalzellen im mütterlichen Kreislauf wurden bei Präeklampsie/HELLP-Syndrom nachgewiesen) eine Rolle spielen. Bei 10 – 20 % der Patientinnen mit ASFL beruhen die Fettsäureoxidationsdefekte auf einer genetischen Prädisposition. Bisher sind fast 20 enzymatische Defekte entlang der mitochondrial lokalisierten Endstrecke der Fettsäureoxidation bekannt, die sich nahezu immer im frühen Kindesalter mit schweren metabolischen Krisen manifestieren. Defekte der Langketten-3-Hydroxyacyl-CoA-Dehydrogenase (LCHAD) gehören zu den häufigeren Störungen der Beta-Oxidation, die zu vermehrter Akkumulation und Exkretion von mittel- und langkettigen Fettsäuren führen. Seit etwa 15 Jahren ist dokumentiert, dass ein fetaler LCHAD-Mangel bei heterozygoter Mutter mit dem Auftreten von ASFL, aber auch von HELLP-Syndrom assoziiert ist (79, 145, 152, 155, 188). Pathogenetisch bedeutsam können hier die vermehrte Produktion toxischer Fettsäuremetaboliten durch den homozygoten Feten, die verminderte LCHAD-Aktivität der Mutter sowie ein sekundärer Carnitinmangel der Mutter sein. Da offensichtlich genetische Faktoren nur bei einem kleineren Teil der Patientinnen eine führende Bedeutung haben, dürften erworbene Faktoren in der Pathogenese der ASFL die Hauptrolle spielen. Unter dem Einfluss von Medikamenten, Ernährungsfaktoren (z. B. Mangel an Riboflavin, Carnitin, essentiellen Aminosäuren), Infektionen (gelegentlich gehen der ASFL virusbedingte Atemwegsinfektionen voraus), Autoimmunreaktionen ausgelöst durch den Feten, genetischer Prädisposition sowie der hormonellen Umstellung in der Schwangerschaft, Faktoren, die in variablen Kombinationen wirksam werden können, ist für die ASFL eine multifaktorielle Genese anzunehmen. Weitere, die Beta-Oxidation hemmende und noch zu definierende Stressoren sind denkbar. Klinik. Wie beim ALV aus anderer Ursache beginnt auch die ASFL mit einer primär unspezifischen Symptomatik. Die bis dahin meist völlig gesunde Schwangere klagt über Krankheitszeichen wie Oberbauch- oder Rückenschmerzen, Appetitlosigkeit, Übelkeit, kaffeesatzartigem Erbrechen, Müdigkeit und Kopfschmerzen, die sich in den nächsten Tagen zunehmend verstärken. Gleichzeitig beobachtet man eine Tachykardie mit einer Frequenz von 100 – 160 pro Minute, ohne dass ein Schockzustand, eine Herzerkrankung, Fieber oder eine Anämie vorliegen. In der Regel entwickelt sich in den nächsten Tagen ein rasch zunehmender Ikterus. Die Kranke wird zunehmend komatös und weist die verschiedensten neurologisch-psychiatrischen Krankheitserscheinungen auf. In dieser Krankheitsphase können jedoch auch andere Symptome klinisch in den Vordergrund treten, die Ausdruck von Störungen weiterer Organe sind, und die die akute Schwangerschaftsfettleber als Multisystemerkrankung ausweisen (Tab. 1.18) (69, 70, 74, 151). Hepatische Enzephalopathie. Die hepatische Enzephalopathie ist auch bei der ASFL das Kardinalsymptom, dessen Pathogenese multifaktoriell und noch nicht völlig geklärt ist. Bei drei Vierteln der bisherigen Beobachtungen wurden die verschiedensten psychischen und neurologischen Störungen beschrieben, ein Koma als schwerste Veränderung entwickelten 40 %. Allerdings stützen sich derartige Aussagen auf den subjektiven Eindruck der einzelnen Autoren in den bisherigen Publikationen, eine Objektivierung wie die Einteilung der Enzephalopathie in 4 Schweregrade wurde bisher nicht vorgenommen. Innerhalb weniger Stunden kann sich die Enzephalopathie von Grad 1 zu Grad 4 fortentwickeln, parallel zu den Schweregraden steigt das Risiko von Hirnödem und Multiorganversagen. Klinische Zeichen des Hirnödems im Rahmen einer Enzephalopathie Grad 4 sind u. a. systolischer Blutdruckanstieg, Hyperventilation, Tachykardie, Krampfanfälle und schließlich ein Ausfall der Hirnstammreflexe. Generalisierte Krampfanfälle deuten bereits auf einen irreversiblen Hirndruck hin, fast alle der bisher beobachteten Schwangeren kamen ad exitum. Tab. 1.18 Symptome und Komplikationen der ASFL
Kardiovaskuläre Störungen. Physiologisch führen in der Schwangerschaft hormonelle Umstellungen zu einer hyperdynamen Zirkulation mit Anstieg von Schlagvolumen, Herzminutenvolumen und Herzfrequenz sowie zu einem Abfall von Gefäßwiderstand und Blutdruck. Die gleiche hypotone, hyperdyname Kreislaufsituation findet sich beim ALV wie auch bei Patienten mit Sepsis. Entsprechend sind die hämodynamischen Veränderungen besonders stark bei der ASFL ausgeprägt und führen zu Störungen von Mikrozirkulation und Gewebeperfusion. Lokale Laktatazidosen und eine allgemeine metabolische Azidose sind die Folge. Pathophysiologisch kommen Hypotonie bis hin zum Schock, Mikrozirkulationsstörungen und Sepsis eine große Bedeutung in der Entwicklung des Multiorganversagens zu, entsprechend stellen diese Faktoren häufige Todesursachen dar. Etwa ein Drittel der Patientinnen mit ASFL wiesen Symptome einer Präeklampsie auf. Hypertone Blutdruckwerte finden sich in 30 % der Fälle, die Werte liegen überwiegend im Bereich zwischen 160/95 mm Hg und 200/110 mm Hg. Auf den systolischen Blutdruckanstieg bei ALV und Enzephalopathie Grad 4 als Folge einer ödembedingten intrakraniellen Druckerhöhung wurde bereits hingewiesen. Gerinnungsstörungen. Komplexe Gerinnungsstörungen kennzeichnen ebenfalls das ALV, bedingt durch eine verminderte Synthese in der Leber und einen gesteigerten peripheren Verbrauch. Es besteht ein Mangel an: Fibrinogen, Prothrombin, Faktoren II, V, VII, IX, Protein S, Protein C und AT III. Der Quick-Wert (Prothrombinzeit) ist erniedrigt. Häufig liegt gleichzeitig eine Thrombozytopenie und Thrombozytopathie vor. Eine latente Verbrauchskoagulopathie liegt bei der ASFL praktisch immer vor, das Vollbild ist dagegen selten. Die hämorrhagische Diathese führt bei zwei Dritteln der ASFL-Patientinnen zu Blutungen unterschiedlicher Art, wie Blutungen in die Haut und Schleimhäute sowie genitale Blutungen unter der Geburt. Gastrointestinale Blutungen sind häufig mitverantwortlich für den letalen Ausgang. Sie stammen aus akuten Erosionen und Ulzerationen von Ösophagus, Magen und Duodenum, selten aus Jejunum, Ileum und Colon. In Einzelfällen wurden auch Blutungen aus Mallory-Weiss-Läsionen oder aus Ösophagusvarizen beschrieben. Offenbar kann hier die physiologische portale Schwangerschaftshypertension transitorisch einen weiteren Druckanstieg erfahren durch die sinusoidale Kompression infolge der diffusen Leberzellverfettung. Infolge der Gerinnungsstörungen können sich auch Leberhämatome ausbilden, sogar über eine Leberruptur wurde berichtet (120). Akutes Nierenversagen, Polydipsie-Polyurie-Syndrom. Ca. 80 % der Schwangeren mit ASFL entwickeln eine rasch progrediente Einschränkung der Nierenfunktion mit Oligurie und Kreatininanstieg, Proteinurie und Abfall der Natrium-Ausscheidung im Urin, gefolgt von akuter tubulärer Schädigung mit Anurie. Ursachen dieses prärenalen akuten Nierenversagens sind eine akute Hypoperfusion infolge renaler Vasokonstriktion, Sepsis und Volumenmangel (mangelnde Flüssigkeitszufuhr, gastrointestinale Blutungen, Emesis, akute Pankreatitis, Fieber, Aszites, Ödeme) mit anschließendem hypoxischen Tubulusschaden. Es ist unklar, ob die ähnlichen mikrovesikulären Verfettungen, die sich wie in der Leber auch in renalen Tubulusepithelien finden können, funktionell von Bedeutung sind. Unter adäquater Therapie ist das Nierenversagen lange Zeit reversibel. Im Stadium der Oligoanurie ist besonders auf das Auftreten von Überwässerung (Lungen- und Hirnödem), Hyperkaliämie (Herzrhythmusstörungen) und metabolischer Azidose zu achten. Einige Patientinnen mit ASFL entwickelten in der ersten postpartalen Woche ein Polydipsie-Polyurie-Syndrom mit täglichen Urinmengen von 7 bis über 9 Litern. Die gleiche Symptomatik wurde auch beim HELLP-Syndrom beobachtet. Es ist noch unklar, ob dieses Syndrom ausschließlich im Rahmen der polyurischen Phase des akuten Nierenversagens zu sehen ist oder ob hier ein zentraler und/oder renaler Diabetes insipidus als eine weitere Komplikation zur Beobachtung kommt. Dies umso mehr, als in der Frühphase der Erkrankung einige Fälle zur Beobachtung kamen, die eine Polydipsie ohne Polyurie aufwiesen, ohne dass man dies z. B. durch die Menge des Erbrochenen ausreichend erklären konnte. Auch in zwei eigenen Beobachtungen trat ein erhöhtes Trinkverlangen von 3 – 4 Litern pro Tag in der Frühphase der ASFL auf (151). Metabolische Störungen. Etwa drei Viertel der bisher beobachteten Schwangeren mit ASFL entwickelten schwere protrahierte Hypoglykämien (Blutzucker < 60 mg/dl). Einerseits können Hypoglykämien zur Verschlechterung der hepatischen Enzephalopathie beitragen, andererseits vermag aber auch die Enzephalopathie die Hypoglykämie zu maskieren. Ursachen der Hypoglykämie sind u. a. fehlende Glykogendepots bei alimentärem Mangel, eine gestörte Glukoneogenese infolge Einschränkung der Leberfunktion und ein Hyperinsulinismus durch verminderte hepatische Extraktion (72). Unter den Störungen des Säure-Basen-Haushalts überwiegt die Alkalose, bedingt durch ein Versagen der Harnstoffsynthese in der Leber mit Akkumulation der Vorläufersubstrate Bikarbonat und Ammonium, häufig assoziiert mit einer Hypokaliämie. Anfangs kann eine respiratorische Alkalose durch Hyperventilation bei respiratorischer Insuffizienz hinzutreten. In fortgeschrittenen Stadien kann sich dann eine Azidose entwickeln. Meistens liegen Laktatazidosen vor aufgrund von Mikrozirkulationsstörungen, Mikrothromben bei DIC und Gewebeödem infolge erhöhter Kapillarpermeabilität. In fortgeschrittenen Stadien mit höhergradiger Enzephalopathie besteht oftmals eine respiratorische Insuffizienz. Für die arterielle Hypoxämie kommen vielfältige Ursachen wie bronchopulmonale Infektionen, Mikrothrombosen, interstitielles Ödem, Kapillarleck mit Lungenödem sowie intrapulmonale Vasodilatation mit funktionellem Rechts-Links-Shunt in Frage. Letztlich können die diversen Schädigungen, die im Rahmen der ASFL auftreten können (Schock jeglicher Genese, Pneumonie, Sepsis, DIC, akute Pankreatitis etc.) eine diffuse, alveoläre Kapillarmembranschädigung (ARDS) als einheitliche Reaktion der Lunge hervorrufen. Das Pankreas ist bei der ASFL (Verfettungen können auch in den Azinuszellen nachweisbar sein) bei verschiedenen Schockformen oder bei Sepsis in unterschiedlicher Häufigkeit beteiligt. Auf der einen Seite finden sich Erhöhungen der Pankreas-Enzymaktivitäten im Serum oder eine ödematöse Pankreatitis als Folge einer Ischämie bei fehlenden oder nicht-führenden klinischen Symptomen, auf der anderen Seite kann sich (in 7 % der Fälle) über eine jetzt gestörte Makro- und Mikrozirkulation eine nekrotisierende Pankreatitis entwickeln. Es ist vorstellbar, dass die nekrotisierende Pankreatitis über eine Freisetzung proinflammatorischer Mediatoren das Schockgeschehen im Gesamtorganismus verstärkt (151). Ein ausgeprägter, im Rahmen der körperlichen Untersuchung nachweisbarer Aszites findet sich bei 16 % der Schwangeren. Als Ursache kommen u. a. ein erhöhter sinusoidaler Widerstand als Folge der akuten Fettleber, eine Hypalbuminämie (verminderte Proteinsynthese in der Leber, vermehrter intestinaler Eiweißverlust) und eine akute Pankreatitis in Frage (151). Beim fulminanten Leberversagen der ASFL ist die Beeinträchtigung wesentlicher Abwehrfunktionen wie gestörte Neutrophilen- und Kupferzellfunktion sowie Mangel an Opsoninen auslösendes Element für das vermehrte Auftreten von Infektionen. Hier sind peripartale und puerperale Infektionen, Wund-, Harnwegs- und Katheterinfektionen sowie Pneumonien zu nennen. Die schwere Sepsis und der septische Schock zählen mit zu den Haupttodesursachen der ASFL, die Anzahl und das Ausmaß der assoziierten Organdysfunktionen beeinflussen dabei wesentlich den Verlauf und die Mortalität der Sepsis. Klinisch-chemische Parameter. Die klinisch-chemischen Parameter bei der ASFL zeigen aufgrund der Beteiligung verschiedener Organe vielfältige Abweichungen. Der Ikterus, oft erst bei Geburtsbeginn auftretend, weist in den meisten Fällen Bilirubinwerte unter 15 mg/dl auf, nur selten werden Werte zwischen 20 und 30 mg/dl erreicht. Aufgrund der hepatozellulären Schädigung ist vorwiegend das direkt reagierende Bilirubin erhöht, ausgenommen die Fälle, in denen eine Hämolyse in den Vordergrund tritt. Hier sind dann die Aktivitäten der LDH und HBDH im Serum entsprechend erhöht und die Haptoglobinkonzentration vermindert (Abb. 1.26). Darüber hinaus kann bei schweren Hämolysen der Wert des freien Hämoglobins erhöht sein. In über 90 % der Fälle sind die Aktivitäten der beiden Transaminasen im Serum erhöht, diese Erhöhungen sind aber als Ausdruck fehlender oder geringer nekrotisierender Prozesse fast durchweg moderat und liegen meist unter 100 U/l, meist sogar unter 50 U/l. Das gleiche gilt für die Aktivität der GLDH. Im Verlaufe der Erkrankung können Medikamente, parenterale Ernährung, Hypoxämie, Hypotension und Sepsis die hepatozelluläre Schädigung verstärken, so dass jetzt cholestatische wie hepatitisähnliche Bilder, relativ häufig auch Mischbilder, beobachtet werden (Abb. 1.27) (77).
Abb. 1.26 Akute Schwangerschaftsfettleber (23-jährige 1-grav., Erkrankungsbeginn 32. SSW, gemini mortui am 17. Tag). Verlauf des Bilirubins und der LDH-Aktivität im Serum. Hämolyse mit Abfall des Hb auf 6,2 g/dl und Anstieg der LDH-Aktivität bis 3000 U/l (HBDH bis 1500 U/l).
Abb. 1.27 Akute Schwangerschaftsfettleber
(dieselbe Patientin wie in Abb. 1.26). Bei den verschiedenen Schockformen zeigt sich ein steiler Anstieg der Aktivitäten von GOT, GPT, LDH und GLDH im Serum (bis zu 1000 U/l und höher). Die Aktivität der γ-GT im Serum ist zunächst normal oder selten leicht erhöht. Allerdings kann es auch hier im weiteren Verlauf vor allem durch medikamentös- oder infektiös-toxische Ursachen zu einer Erhöhung kommen. Als Folge der Synthesestörung kann die Aktivität der CHE auf extrem niedrige Werte unter 500 U/l absinken. Die elektrophoretische Auftrennung der Eiweißfraktionen im Serum kann eine Verminderung des Albumins und eine Vermehrung der γ-Globuline ergeben (Abb. 1.28). Neben der Albuminkonzentration sollte als weiterer Parameter der Leberfunktion Plasmaammoniak erfasst werden. Komplexe Gerinnungsstörungen und Thrombozytopenien prägen das klinische Bild der ASFL. Der Gerinnungsstatus wie die Thrombozytenzahl sind daher regelmäßig zu bestimmen, wie auch Parameter, die über Schäden an extrahepatischen Organen Auskunft geben: Elektrolyte in Serum und Urin, Blutgase, Laktat, Säure-Basen-Status, Blutzucker, Kreatinin, Harnstoff, Harnsäure sowie Lipase und Amylase im Serum (Abb. 1.29 und 1.30). Das Blutbild kann auch ohne Temperaturerhöhung durch eine ausgeprägte reaktive Leukozytose charakterisiert sein. Die Werte bewegen sich meist zwischen 10000 und 30000 Leukozyten pro Kubikmillimeter, aber auch Leukozytenzahlen über 50000 pro Kubikmillimeter sind bekannt. Zum Bild gehört auch das Auftreten von Normoblasten (69, 70, 72, 74, 151).
Abb. 1.28 Akute Schwangerschaftsfettleber (dieselbe Patientin wie in Abb. 1.26) Verlauf der Proteinkonzentrationen und CHE-Aktivitäten im Serum, Gabe von Humanalbumin und Frischblut.
Abb. 1.29 Akute Schwangerschaftsfettleber
(dieselbe Patientin wie in Abb. 1.26)
Abb. 1.30 Akute Schwangerschaftsfettleber (dieselbe Patientin wie in Abb. 1.26) Progredienter Abfall der Thrombozytenzahlen bis zur Geburt (Gemini mortui), Gabe von Thrombozytenkonzentraten. Morphologische Befunde. Die mikrovesikuläre Verfettung der Leber bei der ASFL lässt sich mit bildgebenden Verfahren nicht sicher erfassen, wahrscheinlich bedingt durch die variable Ausprägung der Verfettung. In einer eigenen Beobachtung wiesen nicht mehr als 30 % der Zellen lichtmikroskopisch eine feintropfige Verfettung auf. Bildgebende Verfahren dienen jedoch zum Nachweis von Komplikationen, wie z. B. von Infarkten oder Hämatomen. Die Sicherung der Diagnose ist ausschließlich durch die histologische Untersuchung von Lebergewebe möglich. Es findet sich eine feintropfige Verfettung, die besonders im Läppchenzentrum ausgeprägt ist und die (im Gegensatz zur Tetrazyklin-assoziierten Form, wo sich die Zellverfettung ubiquitär findet) typischerweise um die Portalfelder einen scharf begrenzten schmalen Zellsaum ausspart (Abb. 1.31 und 1.32) Auffällig ist fernerhin, dass diese plurivakuoläre Verfettung unverändert bestehen bleibt; die Fetttröpfchen fließen also nicht wie bei der üblichen Verfettung zu größeren Tropfen oder sogar Fettzysten zusammen, die Leberzellkerne bleiben daher stets zentral gelegen. Die Läppchenarchitektur ist erhalten, nur selten finden sich Einzelzellnekrosen, Gallethromben und kleine portale oder intralobuläre Rundzellinfiltrate. Lag jedoch längere Zeit ein schwerer Schockzustand vor, so können die Läppchen vermehrt mit Einzelzellnekrosen, aber auch mit Gruppennekrosen bis hin zu azinozentralen Massennekrosen durchsetzt sein.
Abb. 1.31 Akute Schwangerschaftsfettleber. Plurivakuoläre Verfettung (40-jährige 5-grav., Sectio caesarea am Aufnahmetag, Tod im akuten Herzversagen). Elektronenoptisch finden sich die zahlreichen kleinen Fetttropfen ohne begrenzende Membran frei im Zytoplasma oder aber auch innerhalb von Phagosomen. Die Mitochondrien weisen häufig Riesenformen auf und zeigen Einschnürungen der Oberfläche. Die Zahl der Cristae ist verringert, kristalline Einschlüsse können auftreten (Abb. 1.33) (69, 70, 151).
Abb. 1.32 Tetrazyklin-induzierte akute Fettleber mit feintropfiger Leberzellverfettung (Prof. Dr. O. Klinge, Kassel).
Abb. 1.33 Ausschnitt einer Leberzelle bei akuter Schwangerschaftsfettleber (23-jährige 1-gravida). Feintropfige Verfettung und weitgehender Glykogenverlust des Parenchyms. Keine strukturellen Veränderungen der Mitochondrien. Vergr. 3180fach Ähnliche Verfettungen wie in der Leber wurden auch in Gastrointestinaltrakt, Pankreas, Niere, Gehirn und Knochenmark gesehen. Makroskopisch ist die Leber bei der Autopsie bei variabler Größe (650 g bis 2350 g in den bisher beschriebenen Fällen) von teigig-weicher Konsistenz und gelblicher Farbe, dabei auffallend resistent gegenüber autolytischen Veränderungen (Abb. 1.34 und 1.35) (69, 70, 151). Die akute Schwangerschaftsfettleber bietet das klinische Bild einer akuten Lebernekrose. Sie muß daher differentialdiagnostisch vor allem von akut nekrotisierenden Hepatitiden (Hepatitis A bis E, Herpes simplex-Hepatitis, akuter Morbus Wilson, medikamentös-toxische Hepatitis) abgegrenzt werden (Tab. 1.16) (57). Dies gelingt zumindest in der Anfangsphase der Erkrankung durch den Nachweis von normalen oder nur leicht erhöhten Aktivitäten von GOT, GPT und GLDH im Serum. Besonders schwierig ist dagegen die Differentialdiagnose des Vollbildes der Erkrankung zu anderen, mit Ikterus einhergehenden Krankheiten am Ende der Schwangerschaft wie schwere Verlaufsformen des HELLP-Syndroms (die Kombination beider Erkrankungen ist möglich), das hämolytisch-urämische Syndrom oder die thrombotisch-thrombozytopenische Purpura. Die in diesen Fällen die Erkrankung definitiv klärende Leberbiopsie verbietet sich meist wegen der gestörten Hämostase, und auch die transjuguläre Leberbiopsie als Alternativmethode wird nicht immer verfügbar sein. Da die Schwangerschaft bei diesen Multisystemerkrankungen mit großer Wahrscheinlichkeit einen krankheitsauslösenden oder krankheitserhaltenden Faktor darstellt, sollte sie durch sofortige Geburtseinleitung oder Kaiserschnitt unterbrochen werden.
Abb. 1.34 Akute Schwangerschaftsfettleber. Makroskopisches Bild (24-jährige 1-grav., Erkrankungsbeginn 38. SSW. Sectio caesarea am 1. Tag, Tod am 14. Tag bei Pneumonie und Peritonitis).
Abb. 1.35 Akute Schwangerschaftsfettleber. Vergleich der Schnittfläche mit einem gesunden Organ (dieselbe Patientin wie in Abb. 1.31). Therapie und Prognose. Die ASFL ist ein lebensbedrohlicher gynäkologischer und hepatologischer Notfall. Da leichte Verlaufsformen in kürzester Zeit in schwere übergehen können, sollte beim geringsten Verdacht die stationäre Behandlung erfolgen, vorzugsweise in einer geburtshilflichen Schwerpunktklinik oder einem Perinatalzentrum in Kooperation mit einem hepatologischen Zentrum. Wie beim HELLP-Syndrom gibt es auch hier keine prädiktiven Parameter, die über Verlauf und Prognose der Erkrankung Auskunft geben. Die Therapie beruht bisher auf zwei Prinzipien: auf der sofortigen Beendigung der Schwangerschaft als auslösendem Faktor und auf der Intensivtherapie des akuten Leberversagens. Da gerade schwere Verlaufsformen von ASFL und HELLP-Syndrom klinisch nicht sicher zu unterscheiden sind, sollte auch ohne Vorliegen eines bioptischen Leberbefundes allein aufgrund des klinischen Verdachts in diesen Situationen eine rasche Schwangerschaftsbeendigung (gegebenenfalls durch Sectio caesarea) angestrebt werden. Die Beendigung der Schwangerschaft bei der ASFL dürfte neben anderen Effekten auch zu einer unmittelbaren Entlastung der pathologischen kardiovaskulären Situation beitragen, da die schwangerschaftsbedingten hämodynamischen Veränderungen sich innerhalb weniger Stunden nach der Entbindung normalisieren. Die Auffassung, bei „milden“ Verlaufsformen der ASFL (eine solche Diagnose ist nur durch die Leberbiopsie zu verifizieren) speziell vor der 34. Schwangerschaftswoche bei unreifem Kind eine expektative Vorgehensweise mit Induktion der fetalen Lungenreife zu erwägen, ist kritisch zu sehen, zumal der Verlauf wie beim HELLP-Syndrom unkalkulierbar ist. Hier ist unter intensivmedizinischer Überwachung in Abhängigkeit vom Krankheitsverlauf, der Stabilität des mütterlichen Zustands und dem Befinden des Kindes in utero individuell zu entscheiden. Die zur Aufrechterhaltung des Kreislaufs und der Vitalfunktionen notwendigen intensivmedizinischen Maßnahmen unterscheiden sich bei der ASFL nicht von anderen Formen des Leberversagens. Eine ausführliche Besprechung der möglichen Therapieoptionen übersteigt den vorliegenden Rahmen. Die in früherer Zeit noch unzureichenden diagnostischen und therapeutischen Möglichkeiten gingen mit einer erhöhten Letalität für Mütter und Kinder einher. Relativ rasch entwickelte sich das durch ein Multiorganversagen gekennzeichnete Vollbild der Erkrankung, das vom ersten Einsetzen der Symptome bis zum Tod der Mutter durchschnittlich etwa 11 Tage dauerte. Der kürzeste Verlauf war 2 Tage, der längste 6 Wochen. Relativ häufig erfolgte 3 bis 4 Tage vor dem Tod der Mutter noch die vorzeitige Spontangeburt eines meist intrauterin bereits abgestorbenen Kindes. Dagegen variierte die Krankheitsdauer bei den überlebenden Müttern zwischen 2 und 8 Wochen und lag im Mittel bei 4 Wochen (69, 70, 151). Die erhöhte Letalität war meist die Folge von hepatischer Enzephalopathie und Hirnödem, akutem Nierenversagen, schweren Infektionen, akutem kardiovaskulären Versagen und Blutungskomplikationen, Komplikationen, die heute gleichfalls die häufigsten Todesursachen bei einem ALV sind. In den vergangenen Jahrzehnten ist es nun als Folge einer raschen Entbindung (bei keiner Patientin hat sich bisher die ASFL spontan vor der Entbindung zurückgebildet) und stetig verbesserter Intensivtherapie gelungen, die mütterliche und parallel dazu die kindliche Sterblichkeit deutlich zu senken (Abb. 1.36 und Abb. 1.37). Schreitet das Multiorganversagen nach der Entbindung fort, sollte vor Eintritt irreversibler Schäden die Indikation zur Lebertransplantation unter Berücksichtigung spezieller Prognosescores diskutiert werden (132). Die mütterliche Letalität liegt jetzt weltweit deutlich unter 20 % und die des Feten unter 30 % (74, 151).
Abb. 1.36 Mütterliche Überlebensrate in 245 Schwangerschaften mit bioptisch gesicherter ASFL.
Abb. 1.37 Kindliche Überlebensrate in 240 Schwangerschaften mit bioptisch gesicherter ASFL. Bei Überstehen der ASFL erfährt die mütterliche Leber eine Restitutio ad integrum, am ehesten sind renale oder zerebrale Residuen zu befürchten. Das Rezidivrisiko einer ASFL bei erneuten Schwangerschaften ist gering, ausgenommen die Fälle, in denen LCHAD-Defekte vorliegen. Hier liegt das Risiko aufgrund des autosomal- rezessiven Vererbungsganges bei 15 bis 25 %. Phäno- und genotypisch sollte daher jedes Neugeborene auf LCHAD-Defekte kontrolliert werden. Es empfiehlt sich, auch den Vater in die Untersuchung einzubeziehen. 1.4. Literatur
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||